-
Neuroscience & Pharmacology Discussion Welcome Guest
Posting Rules Bluelight Rules Recent Journal Articles Chemistry Mega-Thread FREE Chemistry Databases! Self-Education Guide -
N&PD Moderators: Skorpio
You are using an out of date browser. It may not display this or other websites correctly.
You should upgrade or use an alternative browser.Novel Opioids
- Thread starter Feretile
- Start date




Fertile
Bluelighter
- Joined
- Mar 31, 2022
- Messages
- 1,627
The above is a fragment of the morphine molecule, specifically the A,B & D rings.
Now it's too complex to be worthwhile synthetic production... but like morphine analogues, the N-methyl can be replaced by other groups thus:
N-methyl - 1
N-phenylethyl - 8
N-beta hydroxy phenylethyl - 16
N-(p-aminophenyl)ethyl - 20
N-(2-furanyl)ethyl - 60
N-(2-thienyl)ethyl - 30
So it's reasonable to suggest that if one can get a Chinese fine chemical company to provide the N-desmethyl compound as a precursor and very likely the appropriate aldehydes, it's possible to produce quite legal opioids with a potency around that of fentanyl. It's worth stating that acetylation of the phenol results in LESS potent compounds. But none of the precursors are watched and animal studies suggest that the duration of these compounds are similar to the phenanthacene opioids i.e. the T½ is around 2.5 hours and so I would expect the duration of action to be 4-6 hours.
While I freely admit that this class is more costly to produce than fentanyl derivatives, if someone were prepared to acquire the appropriate machinery to produce high-quality foxycodone (fake oxycodone), the pills would fetch the same prices as authentic OyyContin (which I believe ended up being worth ¢50/mg thus a fake 40mg OxyContin would be worth $30.
I mention this because it is my belief that the people with the resources to do this lack the skills and vice versa so it's never going to turn up on the street.
After all, Z4349 is x312 morphine... and NOBODY has ever produced it.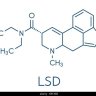
Delmonte421
Bluelighter
- Joined
- Oct 16, 2022
- Messages
- 1,118
why does the S stereoisomer increase activity over the R?I don't think anyone knows or, rather, all they do is test the activity. From the report, beta hydroxy fentanyl is significantly more potent and since it's the (S) that increases activity.... it might well be applicable in other situations.
Fertile
Bluelighter
- Joined
- Mar 31, 2022
- Messages
- 1,627
why does the S stereoisomer increase activity over the R?
Because the phenolic aromatic has to be in the correct relative spatial position so only that 1 isomer has said relative position.
The other isomers are NMDA antagonists, so you might not need to resolve.
Delmonte421
Bluelighter
- Joined
- Oct 16, 2022
- Messages
- 1,118
very interesting, like the well know case of ThalidomideBecause the phenolic aromatic has to be in the correct relative spatial position so only that 1 isomer has said relative position.
The other isomers are NMDA antagonists, so you might not need to resolve.
Fertile
Bluelighter
- Joined
- Mar 31, 2022
- Messages
- 1,627
Well in fact, the makers resolved the 2 isomers of thalidomide and removed the 'inactive' one - but in vivo racemization occurs, so you end up with some of the 'inactive' (read mutagenic) isomer in the body. It's exact action is hotly contested but intercalation into DNA seems likely given it's chiral metabolites.
Generally this doesn't happen, but it wasn't realized that the phthalimide moiety itself is a zwitterion (rapidly switches between 2 forms) which meant an achiral substance.
A true nightmare. I cannot imagine what those families went through.
What IS sad is that one day an RC will turn up that is just as harmful. I mean, with K people HAD to suffer symptoms and go on using, but sooner or later someone will 'design' something with a very specific and undetectable toxicity. It may have happened. We may start to see a lot of deaths... but the longer vendors keep guessing, the more certain this is.
Delmonte421
Bluelighter
- Joined
- Oct 16, 2022
- Messages
- 1,118
Thats the worst part about RC drugs is that you really never know what its going to do to you unless you run clinical trials. It's insane to me the amount of people willing to try drugs on themselves with no working knowledge of med chem or biochemWell in fact, the makers resolved the 2 isomers of thalidomide and removed the 'inactive' one - but in vivo racemization occurs, so you end up with some of the 'inactive' (read mutagenic) isomer in the body. It's exact action is hotly contested but intercalation into DNA seems likely given it's chiral metabolites.
Generally this doesn't happen, but it wasn't realized that the phthalimide moiety itself is a zwitterion (rapidly switches between 2 forms) which meant an achiral substance.
A true nightmare. I cannot imagine what those families went through.
What IS sad is that one day an RC will turn up that is just as harmful. I mean, with K people HAD to suffer symptoms and go on using, but sooner or later someone will 'design' something with a very specific and undetectable toxicity. It may have happened. We may start to see a lot of deaths... but the longer vendors keep guessing, the more certain this is.
Fertile
Bluelighter
- Joined
- Mar 31, 2022
- Messages
- 1,627
J Med Chem 1977 Nov;20(11):1496-9. doi: 10.1021/jm00221a027.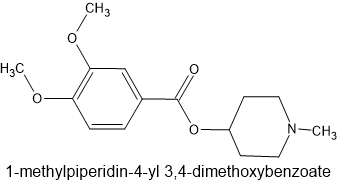
Heterocyclic and piperonylic acid esters of 1-methyl-4-piperidinol as analgesics
Example 23
The above compound is about x2 codeine in potency which gives insights into the structure of such compounds as metofoline (the 3,4-dimethoxy moiety), the phenylpiperidine classes and dimethylaminopivalophenone (ketone replacing quaternary carbon).
I remain convinced that dimethylaminopivalophenone is worth exploring. Further substitutions (such as dimethyl being replaced by other amine) were all less or inactive. The reason for this is simple to explained if one uses Chem3D and overlays it with pethidine, prodine or similar. One of the 2 methyls overlays the N-methyl of the piperidine ring but the other is PART of the piperidine ring.
The obvious first idea is to try using an asymmetrical amine i.e. N-methyl-N-<something else> but when that is done, it's still the methyl that replaces the N-methyl of the phenylpiperidines.
In fact, oddly enough, the core of the mitragynine molecule is the closest since the double-bond between the pyrrole of the indole and the piperidine acts just like the dimethyl moiety of dimethylaminopivalophenone i.e. it makes the compounds more rigid (a key to high affinity).
My personal opinion is that like sibutramine, replacing a dimethyl with a cyclobutyl doesn't alter the spatial position of the alkyl groups on either size. Also it's not much larger but key - it will increase the LogP (another key to high activity).
Fertile
Bluelighter
- Joined
- Mar 31, 2022
- Messages
- 1,627
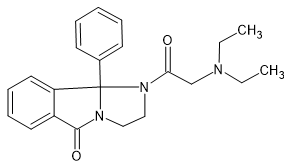
W. Graf, Swiss Patent No. 481,124
This compound possesses analgesic, sedative, antitussive, hypotensive, and spasmolytic properties.
Well the above has features of methadone bezitramide and a few others. Far too complex to make, BUT if it's highly active, it's useful in a QSAR analysis.
Fertile
Bluelighter
- Joined
- Mar 31, 2022
- Messages
- 1,627
3-substitued ketobemidone derivatives.
Ketobemidone is interesting. So far NOBODY has found an analogue that is a pure opioid agonist. Researchers discovered that increasing the length of the N-alkyl to an N-pentyl increases kappa activity so as an analgesic, it's as potent. Dysphoria is the problem. Thus while kwtobemidone is controlled, no analogues are.
Of most interest is the 3-methyl derivative. Otto Eisleb (who also discovered demeraol, MPPP, prodine and ethylprodine) tried adding a 3-methyl which theory states is x7.5 more potent. It wasn't, it was an antagonist.
But their are in fact 4 enantiomers of 3-methyl ketobemidone. I think it likely that some are agonists
and some are antagonists. The clue is Picenadol (LY-97435). It's described as a 'mixed agonist' because 1 isomer is an agonist, another an antagonist. The others have much less activity.
So chiral synthesis or optical resolution might well produce a potent chiral agonist.
Fertile
Bluelighter
- Joined
- Mar 31, 2022
- Messages
- 1,627
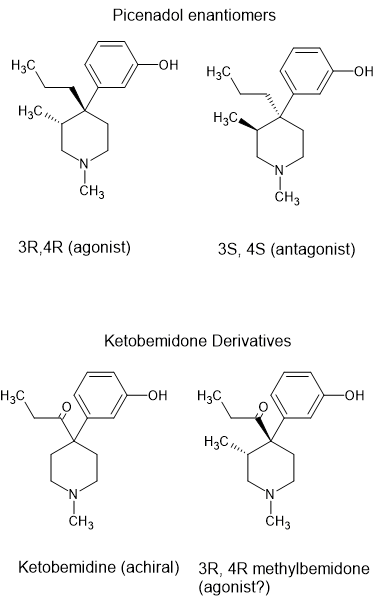
I thought I should draw images so people can follow the logic of the above post.
As far as I can tell, picenadol isn't covered by any specific law but generally speaking, a 3-methyl added to a 4-phenylpiperidine opioid increases potency by an order of magnitude.
The index patent is:
US4081450A '1,3,4-Trisubstituted-4-arylpiperidines and their preparation' by Dennis M. Zimmerman of Eli Lilly
but it's well worth the effort at looking at the patents that cite the above.
In human trials, the racemate is considered to be x4 meperidine in potency. But it's interesting that the synthesis allows the trans isomers to be produced so that only 2 isomers are produced and thus fractional crystalization seems likely. It's digging out that reference.Last edited:
paracelsius
Bluelighter
- Joined
- Mar 11, 2020
- Messages
- 197
what about the phenethyl homolog of ketobemidone has it ever been done? like that of meperidine:
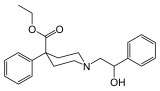
this compound is full agonist about 10xM. apparently, that specific compound (and one with carbamoylethyl instead of phenylethyl) are specifically excluded from Canada controlled substances list as well as UNDOC list. so they're perfectly legal, at least in canada. wonder why they specifically excluded.. that old UN paper claiming "too irritating to test"?? wtf!! did they tried snorting them? kind of stupid if you ask me.
Fertile
Bluelighter
- Joined
- Mar 31, 2022
- Messages
- 1,627
They all seem to be antagonists and/or have significant kappa receptor activity. Many analogues have been produced but I noted that in no case did they produce a chiral target and to resolve the enantiomers to check for the activity of each one.
The simplest was making the N-methyl longer. The most potent was the n-pentyl but it also produced withdrawal sydrome in animals dependend on opioid agonists.
Fertile
Bluelighter
- Joined
- Mar 31, 2022
- Messages
- 1,627
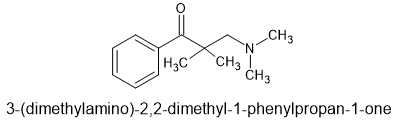
87% yield in 1 step from commercially available chemicals. Cost about £128/Kg i.e. cheapest and simplest opioid known to man.
Fertile
Bluelighter
- Joined
- Mar 31, 2022
- Messages
- 1,627
Fentanyl alternative (X600M with a duration of 6 hours).
3-[2-[2-(Furan-2-yl)ethyl]-2-azabicyclo[3.2.1]octan-5-yl]phenol
3-[2-[2-(Furan-2-yl)ethyl]-2-azabicyclo[3.2.1]octan-5-yl]phenol | C19H23NO2 | CID 20364023 - structure, chemical names, physical and chemical properties, classification, patents, literature, biological activities, safety/hazards/toxicity information, supplier lists, and more. pubchem.ncbi.nlm.nih.gov
pubchem.ncbi.nlm.nih.gov
CA1080705A - Azabicycloalkanes and precursors thereof - Google Patents
Abstract of the Disclosure: Novel substituted azabicycloalkanes of the general formula (A) and methods of preparing the same are disclosed. These compounds and their novel precursors are useful as analgctic agents which exhibit a tendency towards low physical dependence.patents.google.com
Page bookmark CA1080705 (A) - AZABICYCLOALKANES AND PRECURSORS THEREOF
Inventor(s): ONG HELEN H; ANDERSON VERNON B +
Applicant(s): HOECHST AG +
Fertile
Bluelighter
- Joined
- Mar 31, 2022
- Messages
- 1,627
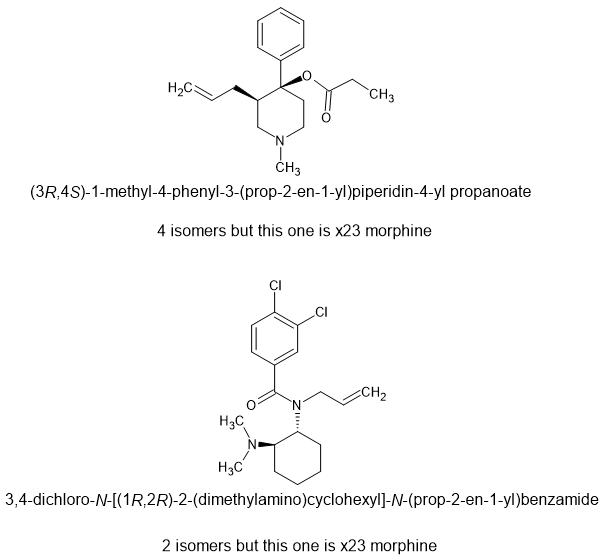
So while U-47700 is x7.5M, U93951 is x12 morphine, isn't controlled and is as cheap to make as it's parent. The allyl moiety binds to an extra amino-acid residue. While Upjohn simply placed emphasison placing the N: in the correct spot - this uses known extra binding regions.
Given how for fentanyl is cut and how rubbish it is, a highly euphoric opioid with a duration of 6 hours should be worth much more and is LEGAL in most nations.
Fertile
Bluelighter
- Joined
- Mar 31, 2022
- Messages
- 1,627
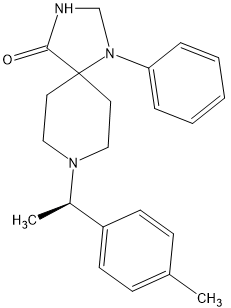
I found the active isomer and discovered that the other isomer had no opioid affinity and so the above is x900 morphine in potency and can be made in 3 steps as the 1-phenyl-1,3,8-triazaspiro[4.5]decan-4-one (which seems like a rare precursor) is in fact used as a precursor to several medicines including Imap, the long-acting neuroleptic often given as a depot medication. Since only 3 manufacturers exists ensure that it is commercially avsailable.
But stronger analogues are known:
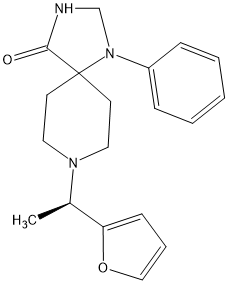
The above is x1100 morphine.
Oh, and in both cases the duration is 8 hours. So 0.25mg TID would prevent a rattle.
Fertile
Bluelighter
- Joined
- Mar 31, 2022
- Messages
- 1,627
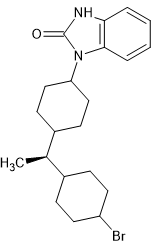
I'm sure prople know of brorphine. It's actually taken from a Chinese people on biased ahonists.

A G protein signaling-biased agonist at the μ-opioid receptor reverses morphine tolerance while preventing morphine withdrawal
It has been demonstrated that opioid agonists that preferentially act at μ-opioid receptors to activate G protein signaling over βarrestin2 recruitment produce antinociception with less respiratory suppression. However, most of the adverse ... www.ncbi.nlm.nih.gov
www.ncbi.nlm.nih.gov
But importantly it shows how the fentanyl moiety can be folded into a smaller space.
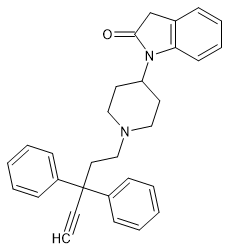
It'd interesting that quite different opioids like bezitramide & benperidol share the1,3-dihydro-2H-benzimidazol-2-one moiety.
What IS worth noting is that neuroleptics and opioids are often very similar in structure.
SR-16435 has a 1,3-dihydro-2H-indol-2-one and so it seems likely that an appropriste N-substituent.