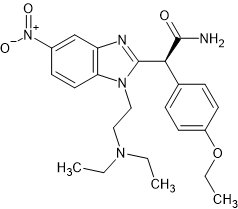
I have been fascinated by the various etonnitazone homologues sold by various dubious Chinese RC vendors. What I find strange is that in 1961 a German team synthesized about 60 homologues and analogues based on intelligent design. It was hardly a stretch to appreciate that a -OH (or -OCH3) on the benzyl spacer overlaid the 6 position of phenanthracene opioids such as etorphine. Now, the problem with the spacer is that it was not stable, one could not just place an -OH (or -OCH3) onto a side-chain because under physiological conditions., it would rapidly racemise. So, using a simple (but I have to say elegant) solution, they introduced a carboxamide moiety. Now, the catrboxamide has a number of advantages, BOTH isomers are equally potent, addition does not significantly lower LogP and being much more stable (and not attracting enzymes that attack the compound), it does not alter the duration of action nor the metabolism of the medicine.
The result was that a compound estimated to be x60 morphine orally was increased to an activity some x150 morphine.
It should be added that whenever a phenol or secondary hydroxyl moiety is encountered, they can almost always be substituted by a carboxamide. The cocaine medication 8CAC used a 3-carboxyamide moiety to significantly increase it's duration of action. As you may know, in the 1970s it was believed that opiate antagonists would blockade the positive effects of cocaine. They did not.
Still, if the 3-phenol of morphine is replaced by a carboxamide, the resulting compound has a duration of action of around 10 hours. The paper on 8CAC even demonstrated a 2-step synthesis for converting a phenol into a carboxamide but the reagents needed to produced the intermediate -F compound are quite unfriendly.
On a personal note, it's possible to replace both the phenol and the ketone moieties of ketobemidone to produce a novel a potent mu agonists. It does not lend itself to large-scale production but it was an interesting experiment.
I post this in the hope that it spurs others into further research.
BTW I need to do some more work but it would appear that an opioid some x60 morphine can be produced from common, commercially available compounds in 1 step using a name reaction (Mannich Condensation). While it's kind of exciting.... anything x60 M would no doubt end up being sold as a powder and end up killing peopl.e
I am very strongly in favour of opioids <10x OR in the form of sublingual tablets (with maybe 2mg in each) because people can use them orally, sublingually, nasally or (for those intent on death) IV. Sadly, no RC vendor cares about their customers sufficiently to go to such efforts. It's a shame.
Image ea hosted on ImgBB

ibb.co




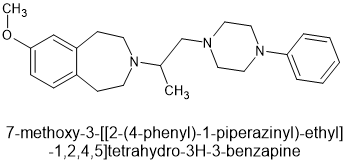
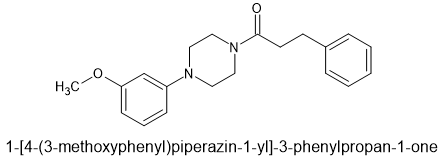
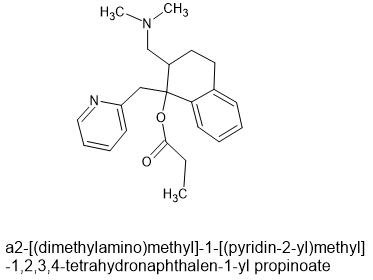
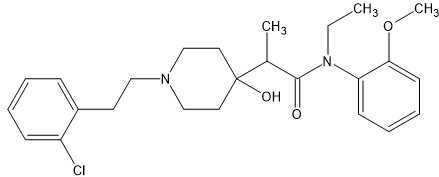

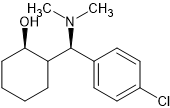
 lone-pairs interact which is hypothesized to be important to bonding. The BEST example is the 6-MeO & beta -OH moiety found in etorphine & related compounds. In the book Opiates (R.Lenz et al), examples of the Bentley compounds lacking the 6-OCH3 absent were tested and potency was reduced from x30o0 to x125 so that N<-->O interaction is important.
lone-pairs interact which is hypothesized to be important to bonding. The BEST example is the 6-MeO & beta -OH moiety found in etorphine & related compounds. In the book Opiates (R.Lenz et al), examples of the Bentley compounds lacking the 6-OCH3 absent were tested and potency was reduced from x30o0 to x125 so that N<-->O interaction is important.