InterestingFACT
Bluelighter
- Joined
- Nov 18, 2013
- Messages
- 512
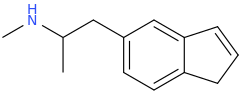
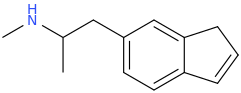
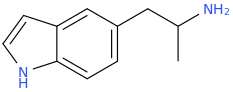
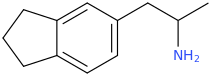
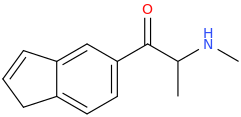
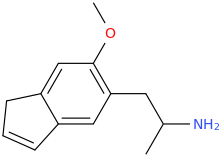
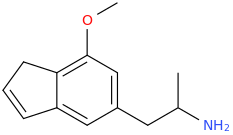
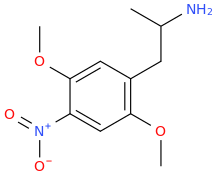
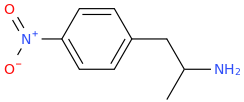
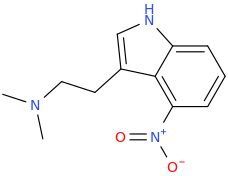
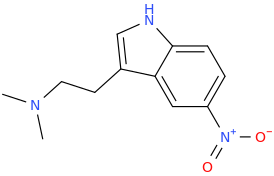
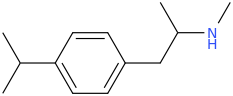
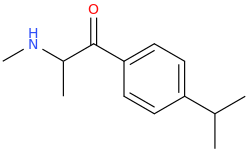
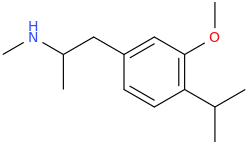
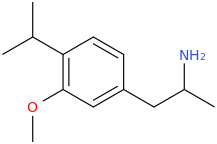
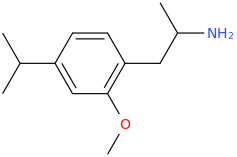
Another oddity which does not seem to fit with the data. Amphetamine and phenmetrazine are known monoamine releasers, with high selectivity for DA and NE. Para fluorine substitution on amphetamine (4-FA) drastically increases SERT affinity. Yet, meta fluorine substitution doesn't increase SERT affinity as expected, but instead slightly reduces it compared to amphetamine. Both phenmetrazine and 3-F-phenmetrazine are very selective for DA and NE but the former compound (ec50 SERT ~8000nM) experiences a 3-4 fold increase from this substitution (3FPM ec50 SERT = 2500nM). What can explain these observations, especially the amphetamine substitution one. SERT tolerates an oxygen meta to the ring (MDMA, 6-APB). Somehow the fluorine is creating slightly negative interactions in 3-FA where as in 4-FA it seems to be binding strongly. The article does suggest a hydrogen bond between one of the fluorines in fenfluramine and the threonine residue, and although the distance will be shorter in fenfluramine, if this is so surely there should be a weaker H-bond in 3-FA and thus would slightly increase affinity rather than decrease it. Any insights are appreciated.
My grasp of chemistry is much looser than many of the folks in this thread, but wouldn't this just have to do with the difference between para and meta substitutions on a benzyl moiety? Halogens are a weird exception in that they're a deactivating group, but they attach ortho/para, against the expected resonance--because they want to dump their lone pair but also end up pulling s electrons from the ring. So a meta halogen is dropping its lone pair on the ring in contrary to the alkyl group's directing, and this would change the resonance therefore either the conformation of the ring, or the partial charge/configuration of the alkyl tail--Which could mean that 3-fa just wouldn't have the right shape.
Last edited: