serotonin2A
Bluelighter
- Joined
- Sep 13, 2014
- Messages
- 1,354
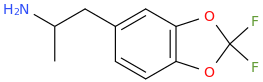
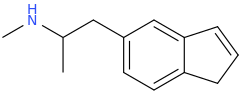
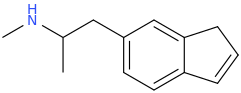
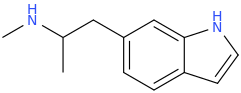
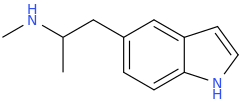
I just recently (thanks to adder) learned that the methylenedioxy ring in MDA is quite out of plane of the benzene ring. What does the indanyl version look like then? Somehow my gut feeling tells that the difference can't be drastic enough to warrant such difference in binding affinity. I would still go with the explanation that the oxygens' electron pairs and/or negative charge have something to do with it.
X-ray crystallographic studies have actually shown that the methylenedioxy ring in MDMA is in the same plane as the benzene ring:
http://onlinelibrary.wiley.com/doi/10.1107/S0108270197012390/abstract
http://www.sciencedirect.com/science/article/pii/S1093326307001908
MM2 energy minimizations on MDA and MDMA confirm that the benzodioxole rings should be in plane.