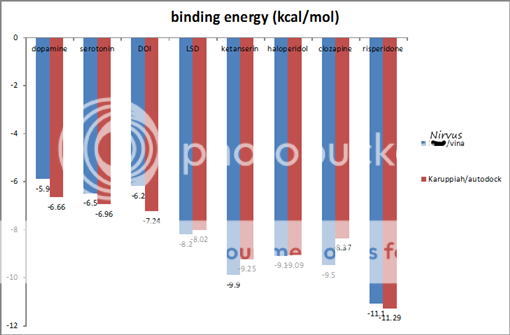
I was learning to use programs that dock small molecules to macromolecules. Then I thought, wouldn't it be nice to have the structure of the 5ht2a receptor! There are so many known (and interesting) ligands: LSD, mescaline, psilocin, serotonin, just about everything in PiHKaL and TihKAL, haloperidol, ketanserin, clozapine, and newer ones: FLY, D-FLY, NBOMe, and TCB2 would all be expected to bind to 5ht2a. Only problem was, the structure for 5ht2a is not publicly available, so I had to make my own. Started with gene sequence for human 5ht2a, ended up with a 3D model that binds known ligands in the appropriate active site, to the amino acid residues that are known to be involved in binding, with binding energies that correlate with published data. In other words, the model is probably very close to the real deal.
So now I can use software to auto-dock molecules to the receptor, and then study the binding interactions, render images, etc. And so I thought I'd share some of the images I've been producing of hallucinogenic 5ht2a agonists interacting with the serotonin 2a receptor. Hope you enjoy!
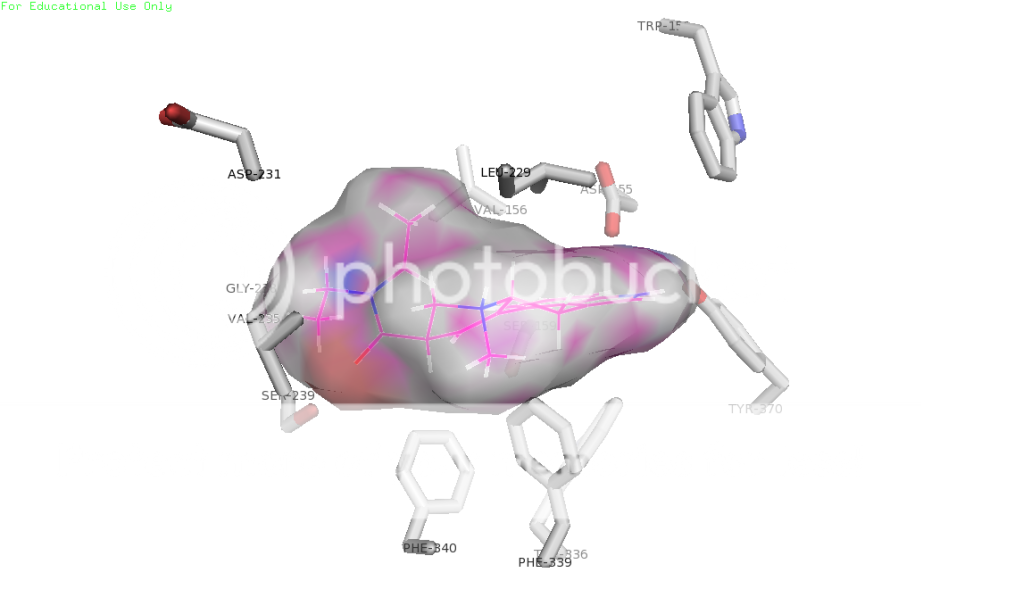
First up, good old lysergic acid diethylamide (LSD-25), shown as space-filling spheres (the balls), bound to the serotonin 2a receptor, shown as a ribbon model.

Stay tuned, more to come. If there are questions, feel free to ask.
So now I can use software to auto-dock molecules to the receptor, and then study the binding interactions, render images, etc. And so I thought I'd share some of the images I've been producing of hallucinogenic 5ht2a agonists interacting with the serotonin 2a receptor. Hope you enjoy!
First up, good old lysergic acid diethylamide (LSD-25), shown as space-filling spheres (the balls), bound to the serotonin 2a receptor, shown as a ribbon model.
Stay tuned, more to come. If there are questions, feel free to ask.

")