www.bluelight.org/xf/threads/what-is-wrong-with-the-mdma-available-today.791073/post-14929097
Aug 21, 2020
There is no doubt in my mind there is a difference. (You nailed it HerpDerpMcDerp; and welcome to Bluelight.)
I’ve been taking MDMA pills and MDMA powder/crystal for 21 years now and after almost giving up, two months ago I had my night rocked by a 120mg dose of the most luscious, munty (of the more cheeky/naughty kind), and long lasting (with God forbid actual afterglow), MDMA crystal in many years. I was on night two as well, having used meth all day prior. Had I been younger, on night #1 and just generally less trashed, this stuff would have been the magic that I remembered from years ago, magic that I have barely encountered in Australia for more than ten years.
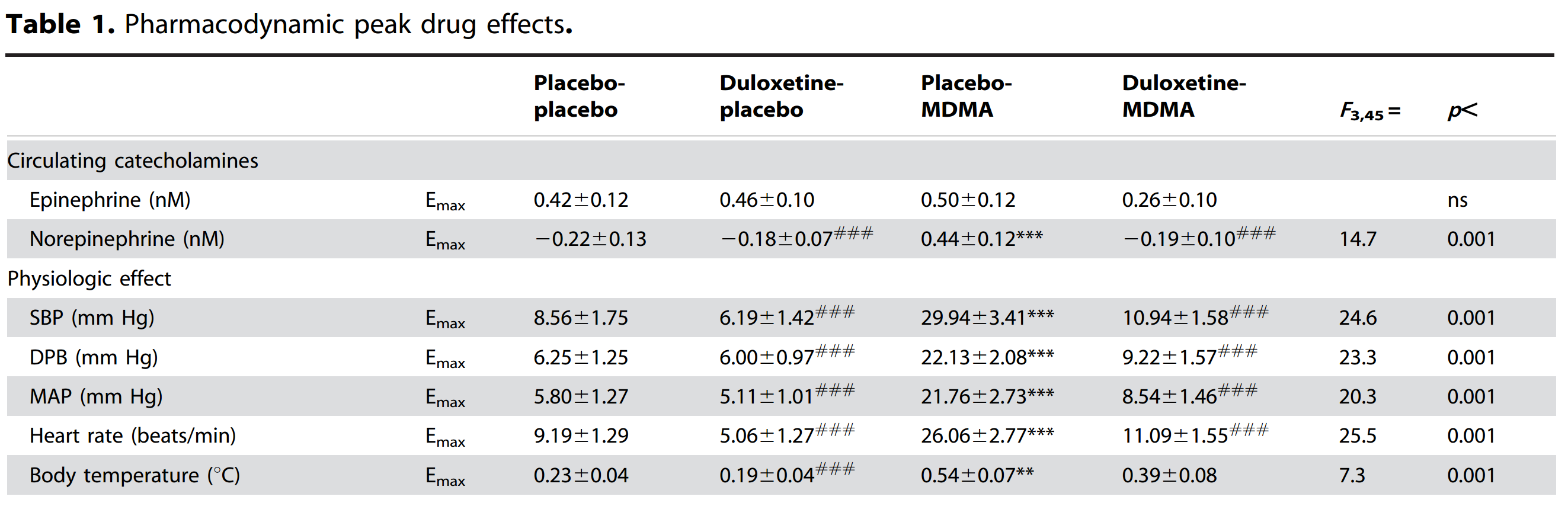
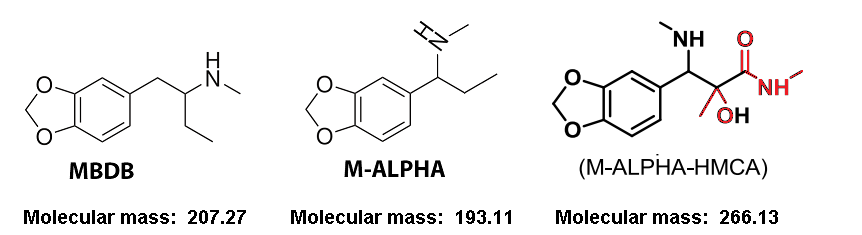
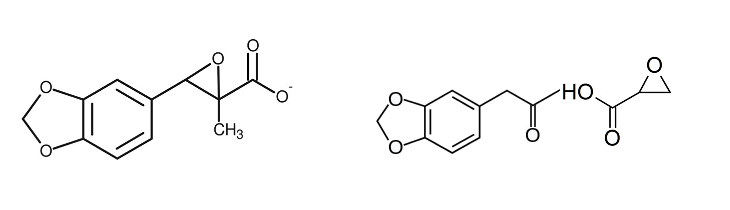
After earlier championing the SvsR isomer issue, I am now positive the answer to this entire issue is contained in the article indigoaura posted many pages ago, where very small quantities of two structurally similar dimers created as impurities during MDMA manufacture, could cause significant inhibition of the MDMA effects at comparatively very low amounts by binding to the neurotransmitter reuptake transporters, much like how for prescription SSRIs and SNRIs nullify the effects of MDMA into something resembling “Meh” in those people taking these drugs. (Didn’t one of the dimers show ridiculously strong affinity to ALL THREE of the significant transporters, the noradrenaline, the dopamine AND the serotonin?! A higher presence of such a chemical in a substance purporting to be “pure MDMA” is going to completely fuck up the roll and will need a much greater dose to get anywhere close to something resembling a roll.)
Whilst I would like to put together a much more thought out, well researched and diagrammatically useful post, as I have been false starting on this endeavour for months now, I thought it better to put something out there for discussion without any further delay.
It is clear that PMK glycidate could favour the creation of these dimers, especially if the PMK is not properly purified. I mean what is created when the glycidate molecule is hydrolysed, acetic acid or acetate perhaps, the very reactant which they list in one of the initial steps that gives rise to these dimers.
The other matter of significance is that of what the limiting reagent is in the reductive animation step of PMK and methylamine Into MDMA. This reaction always ideally proceeds with a significant excess of methylamine to PMK, for if there is no excess, the formation of these dimers or molecules like them is much more likely to occur.
In the old days, as it was safrole/isosafrole and the PMK produced from these that was in the shortest supply, there would never have been any question about having an excess of methylamine to this highly sought after and almost impossible to obtain precursor. However, we know now that PMK glycidates are widely available in massive quantities and so PMK is no longer going to be the limiting reagent in the reductive amination. Instead, not only might manufacturers fail to properly purify the PMK or otherwise proceed via one-pot synth straight to MDMA with potentially highly reactive hydrolysis products from the glycidate (with certain PMK glycidates being worse than others for creating such dimers, this being a further reading for different levels of “meh”), but they may be skimping on the methylamine in the reaction as well, such that there is now an undue excess of PMK made from glycidate relative to the methylamine, a factor which would strongly favour the creation of these inhibiting dimers to a much greater degree. Throw in the fact that platinum catalyst hydrogenation is now almost always used to reduce the imine as opposed to borohydride, and it’s anyone’s guess as to what mess we actually end up with.
The above hypothesis would account for why there are varying degrees of Meh (being the extent to which the MDMA does or does not have the presence of such impurities, a fact which the right laboratory could actually
test for), why safrole produced MDMA doesn’t create this problem to the same degree, and why the change to the almost infinitely available glycidate pre-precursor may have skewed the reaction in the wrong direction on account of commercial considerations (despite the news now of stupidly high dosages of this crappy
MDMA).
As an aside, for a long time high purity racemic methamphetamine produced from p2p has received similarly poor user reports along the lines mehMDMA despite being >90% purity - “mehMeth” if you will. And where is this P2P coming from but BMK glycidates, the PMK glycidate equivalents. Precisely the same reaction considerations to what I have described above for MDMA would apply for methamphetamine produced via this route and where the more readily available precursor might also be present in relative excess to the methylamine needed
for the reductive amination. If similar dimers
were produced (and I’ll find an article I have saved confirming this very thing), then these too would likely inhibit the action of the meth in similar ways.
Sorry that this post is so garbled but the earlier discussion about this and articles i refer to are buried somewhere a hundred pages or so ago. As this thought has otherwise been bubbling uselessly away in my mind for more than six months now, best I just get it down and hope that the more cleverer chemists and those more familiar with the invaluable content in these almost 300 pages, could give it further consideration