cyanide_sunshine
Greenlighter
- Joined
- Apr 26, 2008
- Messages
- 9
PEA is a misnomer to an extent, but bear with me.
I've never tried adderall (or methamphetamine).
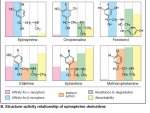
But I certainly have experienced ephedrine and pseudoephedrine, which don't cause alertness, but rather fatigue and high blood pressure. I note these are missing the amine.
Methylphenidate, which has a differing cyclic structure than most PEA, does not cause this in my experience, but has a similar MOA.
I understand this is a little below ADD. As well as anecdotal.
Won't be devastated if no one answers.
I've never tried adderall (or methamphetamine).
But I certainly have experienced ephedrine and pseudoephedrine, which don't cause alertness, but rather fatigue and high blood pressure. I note these are missing the amine.
Methylphenidate, which has a differing cyclic structure than most PEA, does not cause this in my experience, but has a similar MOA.
I understand this is a little below ADD. As well as anecdotal.
Won't be devastated if no one answers.