What you're asking, requires a long response as it's not a black-white answer. So I apologies for that, but that's what you get for being curios
")
What makes a chemical able to be "active" is entirely relative to what you're referring to. I'm going to assume you mean able to cross the blood-brain barrier (BBB). However, the term "active" could apply to any substance that is able to alter bodily function in some way. Therefore, drugs don't have to have an effect on the Central Nervous System in order to be active. They can just as easily have mind altering effects, without crossing BBB.
However, like I said above, I going to assume you mean able to have an effect on the brain. If this is what you mean, my answer is quite different. As far as I'm aware, we don't exactly know what allows a substance to cross the blood-brain barrier. I know that a lot of it has to do with it's electrical charge. Another huge part of what can cross BBB is how soluble in lipids the substance is, and how soluble the substance is in water (but this applies to all solvents in general). Generally - the substance has to be able to be soluble in both (to some degree) to allow it to cross BBB. If it's only soluble in fats, it's not going to cross BBB. If it's only soluble in solvents, it won't cross BBB.
This is known as hydrophilic-lipophilic balance. Hydrophilic-lipophilic balance refers to; if a substance is 100% soluble in lipids (fats), then it cannot be soluble in water (solvents). If a substance is 100% soluble in solvents, then it cannot be soluble in lipids. A substance, therefore, has to have a ratio near 50%/50% solubility in order to cross the blood-brain barrier. (Hope I explained this correctly)
As ebola also said, it also has to do with how "similar" the chemical is to endogenous substances. Although, this is relative to polarity as well. A substance can be exactly the same as a neurotransmitter, but be a different isomer or have a different charge and therefore will not be able to cross BBB.
Methamphetamine is similar to all the monoamine neurotransmitters, not just Dopamine. That's because, Methamphetamine, like all the endogenous monoamines, employs a Phenethylamine skelaton, and therefore is a Phenethylamine derivative, just like the natural chemicals that circle around in your brain meats. (Serotonin isn't as Phenethylamine, but it's pretty damn similar) Just take a look at all of them.
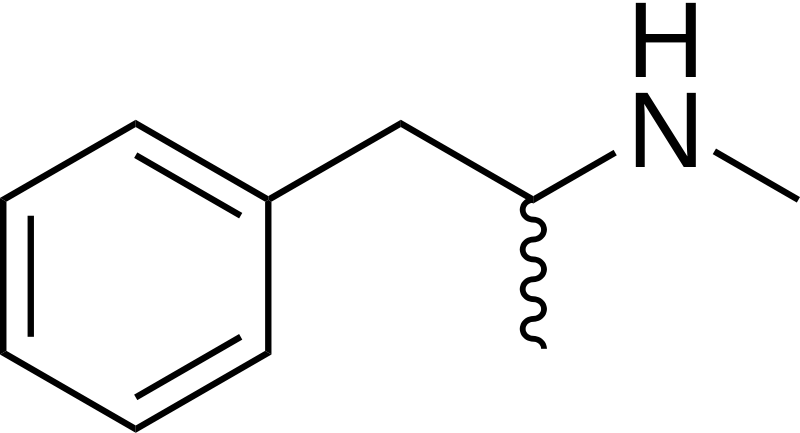
This is Methamphetamine...
Now just compare that to the Monoamines. There pretty damn similar.
Dopamine
Epinephrine
Norepinephrine
Serotonin
Referring back to hydrophilic-lipophilic balance...
Methamphetamine is able to cross the blood-brain barrier far better than Amphetamine. This is due to the fact that Methamphetamine has a more balanced hydrophilic-lipophilic relationship and is, therefore, a "more polar" substance. But what causes this? Well, this would be due to Hydrogen. As someone brilliantly demonstrated in the Amphetamine talk page on Wikipedia....
As you can see in the picture above, Amphetamine has an amide off the chain where Methamphetamine has a Methyl group. Methamphetamine also has three, permanently open hydrogen atoms (meaning permanently open for "bonding") off the bottom chain (I don't know how to refer to these chains at the moment. If someone could enlighten me, that would be great!) Amphetamine, on the other hand, is "less polar" than Methamphetamine due to it's mixed bonding actions, and it's "less polar" amide group.
This allows Methamphetamine to cross the blood-brain barrier far better than Amphetamine. And while Amphetamine does cross BBB, it's under-balanced polarity stops some of it from ever being able to cross the blood-brain barrier.
(Again, hope I explained this correctly. Although, I'm sure someone won't hesitate to correct me if I'm wrong.)