Braenden, Eddy & Halbach (1955), summarizing the relationship between chemical structure and analgesic action, said "In morphine and its derivatives the methyl substituent on nitrogen seems essential because its substitution by other alkyl groups reduces or abolishes analgesic action." It is...

www.unodc.org
Scroll down to the scanned images - these are levorphanol homologues with a variety of N-substituents. The second image lists:
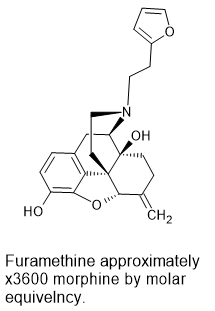
Ro 4-1539 - ED50 0.010 (0.009-0.012) mg/kg
i.e. some x480 morphine in potency.

en.wikipedia.org
Now, their is a simply huge number of antagonists and partial agonists that may be N-dealkylated using non-classical Polonovski reactions (and other methodologies).
N-dealkylation, the removal of an N-alkyl group from an amine, is an important chemical transformation which provides routes for the synthesis of a wide range of pharmaceuticals, agrochemicals, bulk and fine chemicals. N-dealkylation of amines is also ...

www.ncbi.nlm.nih.gov
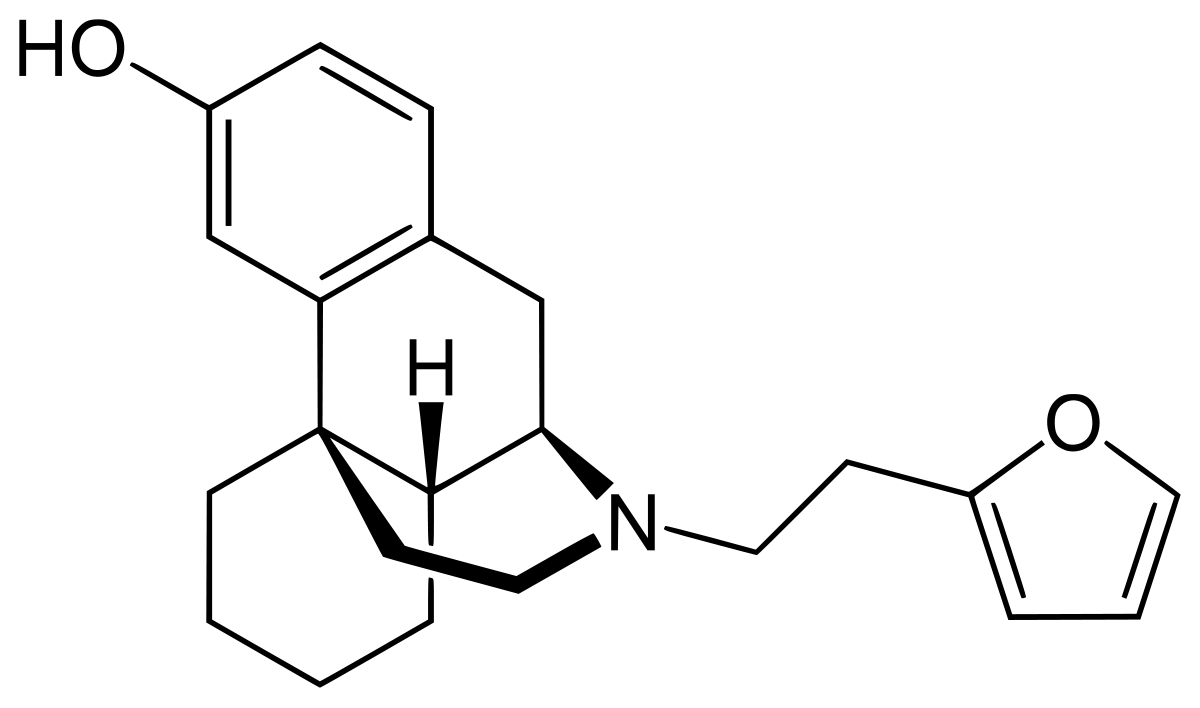
One example would be the N-dealkylation of nalmefene. The N-methyl homologue of nalmefene is some x60 morphine* and so, based on known QSAR data, the N-2-(furan-2-yl)ethyl homologue (like Ro 4-1539) should be around x1600 morphine in potency.
*'Opiates' 1986 by Lenz et all: Page 63 'Structure-Activity Relation of Morphine and Related Structures' Table 3.3
Thus NALMEFENE represents a pre-precursor (2 steps) to:
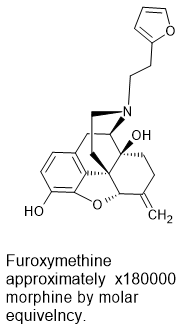
17-(2-(futan-2-yl)ethyl)-4,5α-epoxy-6-methylenemorphinan-3,14-diol
Image 1 hosted in ImgBB

ibb.co
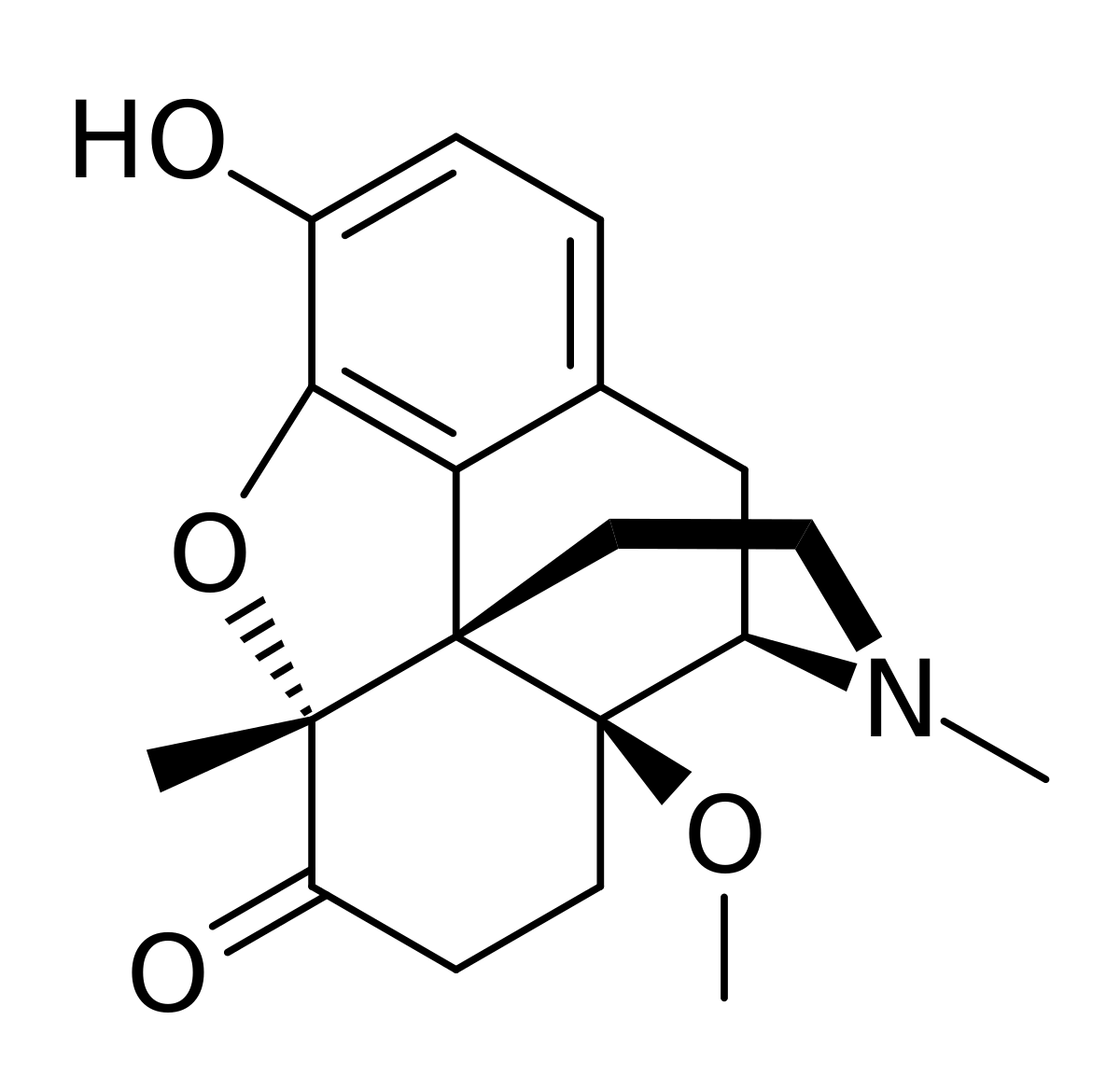
Later work by Professor Helmut Schmidhammer at the University of Innsbruck in the mid-1990s demonstrated that replacing the 14-hydroxy moiety for a 14-methoxy (such as 14-methoxy metopon) increased analgesic activity by almost 2 orders of magnitude. It is believed that this is a result of increased affinity for the DOR.
en.wikipedia.org
So while the QSAR has not been funny explored, it's possible that:
17-(2-(futan-2-yl)ethyl)-4,5α-epoxy-6-methylenemorphinan-14-methoxy-3-hydroxyl
Demonstrates even higher activity but careful examination of his would would be required. But, if it proved to be the case that this compound could in fact be over 100,000 times the activity of morphine. But I would consider a compound with such activity to be a chemical weapon first and foremost:
Image 1 hosted in ImgBB
ibb.co
I should add that a Canadian BLer has already found reference to a compounds that is estimated to be over 100,000x morphine it it's analgesic activity, likely due to possessing both MOR and DOR activity so the above does not represent a range of activity unknown to science.



 bluelight.org
bluelight.org

 pubchem.ncbi.nlm.nih.gov
pubchem.ncbi.nlm.nih.gov