-
N&PD Moderators: Skorpio | thegreenhand
-
Neuroscience & Pharmacology Discussion Welcome Guest
Posting Rules Bluelight Rules Recent Journal Articles Chemistry Mega-Thread FREE Chemistry Databases! Self-Education Guide
You are using an out of date browser. It may not display this or other websites correctly.
You should upgrade or use an alternative browser.
You should upgrade or use an alternative browser.
I Like to Draw Pictures of Random Molecules
- Thread starter nuke
- Start date
- Status
- Not open for further replies.

.. does the site www.chemicalize.org for quick chemical data been jacked? by MoneyGrabbin commercial shit holes..type chemicalize.org and you get redirect to chemicalize.com charging zillions for tax payers public money funded programs.. hack'em
^
It is still in ChemAxon as owner(original), but they seems to manipulatethe site somehow, now i cannot view the site on my ipad anymore(yor browser is not supported, please use desktop version etc etc).
Too bad i visit and utilize the site alot on my ipad
It is still in ChemAxon as owner(original), but they seems to manipulatethe site somehow, now i cannot view the site on my ipad anymore(yor browser is not supported, please use desktop version etc etc).
Too bad i visit and utilize the site alot on my ipad
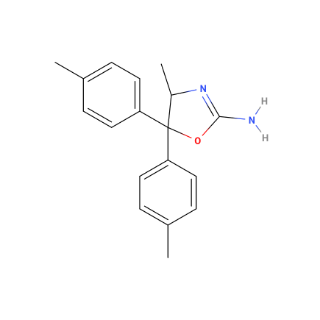
yeah.. too bad! I do use it often too. I guess have to turn to other sources^
Too bad i visit and utilize the site alot on my ipad
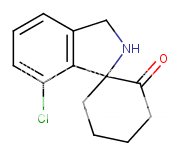
Bagseed
Bluelighter
- Joined
- Jul 22, 2010
- Messages
- 4,042
ring constrained ketamine... does anyone have information on what the best conformation of ketamine would be to fit into the binding site at NMDAr? this constrained version could go either way imho
Yes, hit or miss! But I think it has reasonable chance binding similarly to ketamine. In that case may be 10x more potent than ket because of ring constrained. + it will be legit since it is not an arylcyclohexylamine but an indanyl! The PCP site on NMDA is not particularly demanding: eg (+)DXM ie dextromethorphan and its enantiomer (-)DXM levorphanol both bind NMDAr with similar affinity..It doesn't happen often especially with brain receptors, ions channels..etc. Usually only one enantiomer will bind...this constrained version could go either way imho
The standard NMDAr antagonist, the most potent by far (ca 800x more potent than ketamine) is dizocilpine MK-801 MK-ultra. Very constrained molecule with not many conformations possible: pretty much a single conformation (2 for the racemic!).... does anyone have information on what the best conformation of ketamine would be to fit into the binding site at NMDAr?
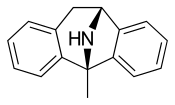
Does arylcyclohexylamines like ketamine, MXE..etc bind at the same site as MK? in that case, overlaying the aryl of arylcyclohexylamines with one of the phenyl of MK and of course the amino groups of the 2. That means adding a phenyl to the cyclohexyl of ketamine MXE..etc(I mean to get something like a tetraline) will give more potent NMDAr than ketamine mxe..etc...and legit! at least in the UK since they won't be arylcyclohexyl but aryltetralinylamines..so technically not cyclohexylamines aryl but who knows?
PS: now MK-ultra is way too potent imho (risks of irreversible permanent pyschosis + Olney's lesions.. but who knows?
Raihiar
Greenlighter
- Joined
- Feb 1, 2011
- Messages
- 43
i didn't go through the whole thread (again), so excuse me if this has already been posted:
since propylhexedrine seems to be at least active, and i was a fan of 4-fa, i just have to ask ^^

i know this "hybrid" or "mixtures" of chemical structures is a bit misleading, you don't get the best of two worlds most of the time..
but it might be active, possibly not very enjoyable, but well... this thread has its name for a reason
since propylhexedrine seems to be at least active, and i was a fan of 4-fa, i just have to ask ^^
i know this "hybrid" or "mixtures" of chemical structures is a bit misleading, you don't get the best of two worlds most of the time..
but it might be active, possibly not very enjoyable, but well... this thread has its name for a reason
aced126
Bluelighter
- Joined
- May 18, 2015
- Messages
- 1,047
i didn't go through the whole thread (again), so excuse me if this has already been posted:
since propylhexedrine seems to be at least active, and i was a fan of 4-fa, i just have to ask ^^
i know this "hybrid" or "mixtures" of chemical structures is a bit misleading, you don't get the best of two worlds most of the time..
but it might be active, possibly not very enjoyable, but well... this thread has its name for a reason
AFAIK the aromatic ring in ampethamine pi stacks with an electron rich tyrosine residue (Tyr 182? Correct me if I'm wrong) in the S1 pocket of DAT which helps stabilise its pose. Losing aromaticity means this interaction is lost, and it will reduce the potency of this drug. Not to mention it'll probably have less affinity for other relevant intracellular proteins like TAAR1 and VMAT. Propylhexedrine is significantly less active than methamphetamine.
Nexus_Tripper
Bluelighter
- Joined
- Nov 16, 2014
- Messages
- 490
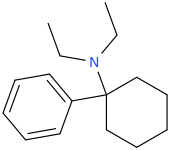
PCDE
Those are junk. Any arycyclohexamin with two things on nitrogen are inert. they are metabolized and one group gets cut of the N, soo if you take enough they can have effects.
Raihiar
Greenlighter
- Joined
- Feb 1, 2011
- Messages
- 43
AFAIK the aromatic ring in ampethamine pi stacks with an electron rich tyrosine residue (Tyr 182? Correct me if I'm wrong) in the S1 pocket of DAT which helps stabilise its pose. Losing aromaticity means this interaction is lost, and it will reduce the potency of this drug. Not to mention it'll probably have less affinity for other relevant intracellular proteins like TAAR1 and VMAT. Propylhexedrine is significantly less active than methamphetamine.
yeah, i know about the huge potency drop, and the same goes for meth vs 4fma, so the already not very potent and side-effect ridden (no 1st hand experience, don't intend to have 1st hand experience) propylhexedrine is probably going to be garbage in the best case and laden withserious side effects - neurotoxicity as with the higher halo-amphetamines, something a la PMA / PMMA or whatever in a worse scenario.. best leave this uncharted land be and sail on :>
Bagseed
Bluelighter
- Joined
- Jul 22, 2010
- Messages
- 4,042

something I drew while on a bus ride recently, no idea if it makes sense, but it looked nice
edit: I am also wondering if a alpha-beta-unsaturated carbonyl is an easy target for metabolism. I made that carbonyl in order to have an sp2 carbon in there to ensure that the ring remained planar.
Last edited:
^ the 4-((2-dimethylamino)ethyl)quinolone work "well" as substitute for DMT: I made similar indoles isosteres back in the days!!Tryptophan analogs with the quinolone replacing the indole ). It might work but lsd doesnt tolerate substitution at 2 position of the indole like with the 2-bromoLSD??) .. but who knows. NB: The one terrible thing about these types of quinolones, they're just terribly insoluble and I mean really pain in the ass to get them into any reasonable solvent.. unless you use boiling DMSO or DMF or .. cold conc sulfuric acid 
). It might work but lsd doesnt tolerate substitution at 2 position of the indole like with the 2-bromoLSD??) .. but who knows. NB: The one terrible thing about these types of quinolones, they're just terribly insoluble and I mean really pain in the ass to get them into any reasonable solvent.. unless you use boiling DMSO or DMF or .. cold conc sulfuric acid 
Nagelfar
Bluelight Crew
- Joined
- Nov 23, 2007
- Messages
- 2,527
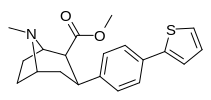
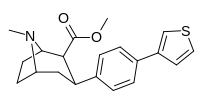
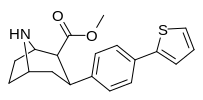
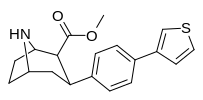
Check this out with regard to the QSAR of phenyltropanes: the top two, 2-para-Thiophene & 3-para-Thiophene have 52 ± 12.8 and 12.1 ± 3 for DAT respectively (i.e. 2-thio has tighter binding for the DAT)
But with the Nor/nitrogen-demethylated of the same compounds, it's the opposite: nor-2-thio is 12.2 ± 0.9 @ DAT and nor-3-thio is twice as potent with 6.4 ± 0.27 @ DAT.... (i.e. the 3-thio is the tighter one)
This shows that the methylation of the nitrogen dramatically changes the position of the benzene substitutions in cocaine like molecules that affects monoamine subtype affinity in drastic ways.
Furthermore, one methylene unit length difference at the nitrogen end seems to correspond to one methylene unit length alteration at the benzene substitution end; see how the middle two compounds have basically the same affinity? But the angles have to be deeper (or shallower, or shall we just say 'more optimized'?), because it has increased affinity as it changes both, whereas just adding another unit again on the same side can completely gimp it.
Last edited:
Bagseed
Bluelighter
- Joined
- Jul 22, 2010
- Messages
- 4,042
interesting, thanks for your comment! maybe it is too bulky, but my feeling says that putting bromine on the 2-position of a Lysergamide will be more bulky than this quinolone thing. bromine is huge! would be interesting to see, if DMT would be active with a 2-bromo-substitution.^ the 4-((2-dimethylamino)ethyl)quinolone work "well" as substitute for DMT: I made similar indoles isosteres back in the days!!Tryptophan analogs with the quinolone replacing the indole
well we'll never know unless someone synthesizes that thing and tries it. where are the shulgins of today?
- Status
- Not open for further replies.