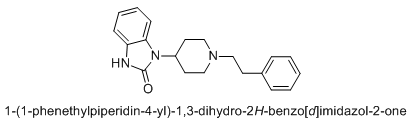
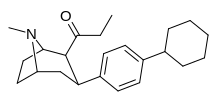
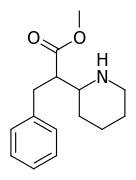
If I were a betting man, I would say that N-methyl-1-(1-phenylcyclohexyl)propan-2-amine and relatives would be poorly active as dissociatives or as stimulants. Sorry Gaffy, nothing personal, but almost every structure you post makes me roll my eyes. The monkeys-at-typewriters approach might yield something eventually but it's not my preferred school of thought.
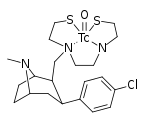
Curiously enough it overlays with a structure that is an isomer of PCP plus a methylene linker. Sort of reminds me of desoxypipradol, so there is still some hope for it having activity. The wacky high LogP and single heteroatom (no metabolic handles) would suggest a compound of rather long duration.
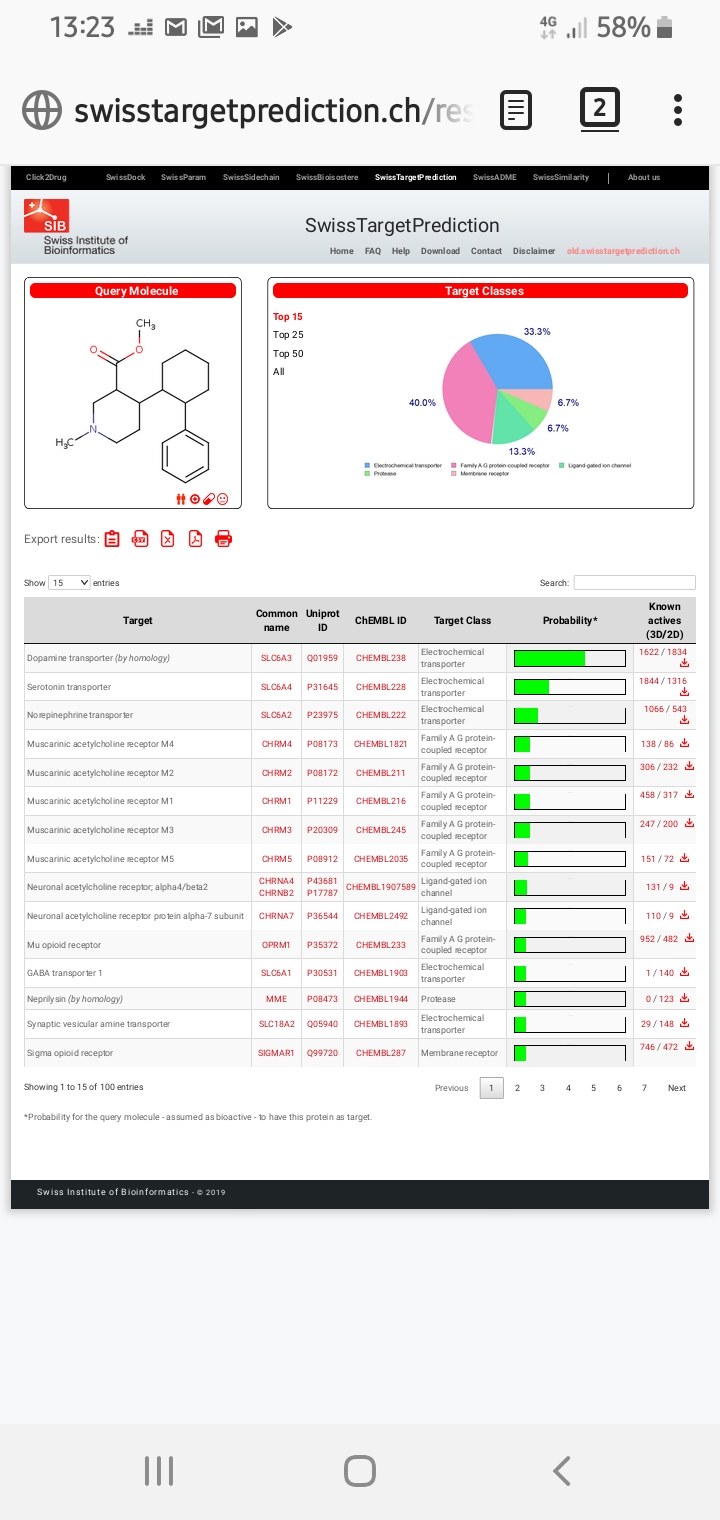
I have to wonder how STP works... does it actually do 3d modelling behind the scenes or is it predicting binding off of strictly 2d structures? Maybe I will have to play with feeding chiral compounds into it and see what changes, if anything. I have a bad feeling that it is a simple machine learning type thing that takes a SMILES/2D structure and matches similarity versus certain known standard compounds. This would explain why it says
(by homology) on some results. Maybe it's doing prinicpal component analysis or something?
If you want a concerning demonstration on how STP is not as accurate as you'd like, loperamide seems to be a scaffold that it has a very hard time with. Lope itself is correctly identified as a mu-ligand, and as I understand it it is reasonably selective for that receptor. Now for some reason, STP says that it binds at 5-HT2b, Histamine H2, Adrenergic beta, SERT, Adrenergic alpha-1a, mu opioid, Dopamine D3, delta opioid, kappa opioid, sigma opioid, CYP2D6, tyrosine kinase TIE-2, Adrenergic alpha-2a, Adrenergic alpha-2b, Dopamine D4, Histamine H1, and Histamine H3, all with very high probability. Last I checked that's 16 extra targets predicted that don't actually factor into loperamide's activity at all.
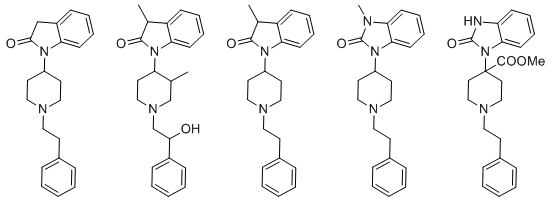
Doing things like adding a 3-methyl (on the piperidine), or swapping dimethylamide to pyrrolidine amide, or substituting the
para-chlorine for another halogen or a methoxy, all things that should result in moderate changes in affinity at mu but very little else, don't help either. It just makes things even weirder.
This all seems to point to STP not doing anything too fancy and certainly not doing any actual modelling or docking simulations.
Nagelfar, would you mind if I asked you to add this one to your methylphenidate analogs?
I think you'd need to track down a paper where it was synthesized and tested before it gets an official entry. I don't like putting words in other people's mouth - but if you look at the carefully curated pages Nagelfar's assembled, they are all backed by peer-reviewed data. (You will notice a definite lack of any STP guesstimates there!) That is, the compounds have actually been made and tested and had their binding affinities measured. That's part of why I find it so impressive and useful.
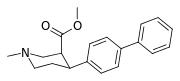
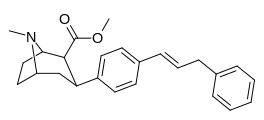
I would like to add that ring-substititions of the phenyl on these PCs seems to make them almost inactive, maybe substitutions makes these bulky compounds "too" bulky? ^^
Or it could just make the compounds different enough from a structural standpoint, decreasing the quality of matches to known actives, and reducing the probability thusly.
")
 swisstargetprediction.ch
swisstargetprediction.ch
 swisstargetprediction.ch
swisstargetprediction.ch