aced126
Bluelighter
- Joined
- May 18, 2015
- Messages
- 1,047
...DA releasing agents? Also, why, on a molecular level, is MDAI less selective for DA neurons?
I'm in doubt over whether the absence of neurotoxicity is related to MDAI's pharmacodynamic profile or if it's something to do with the actual molecule itself.
There is a lot of evidence that a lot of selective SRAs aren't neurotoxic due to the fact that not much DA release occurs and that not much DA can as a result be taken up into 5HT neuron and cause damage from there. For example, when a DRA and MDAI are co-administered, neurotoxicity is observed.
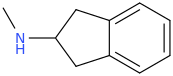
In terms of the molecule, the alpha methyl group is cyclised back onto the benzene ring. The structure is below for reference. I remember reading on wikipedia (can't seem to find on what page I read it, it might have been on the MDAI page itself but taken off for some reason) that because the alpha methyl is cyclised, there is no chance for methyl radicals to form unlike in MDMA, and that this plays a key in preventing cellular damage. I do not know much about radical chemistry at all, so if someone could evaluate these reasonings, that would be appreciated and spur interesting discussion.
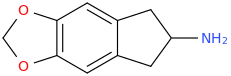
Furthermore, I pondered over the fact that cyclising the molecule back to the ring could so drastically increase it's serotonin-dopamine selectivity (10:1 from wikipedia article). No new functional groups added, only a (small) change in conformation. Could it be that in fact because the molecule is rigidified and cannot rotate around the indane ring, it would always be in a decent conformation to interact with SERT (MDMA interacts better obviously but MDAI and its conformation definitely does interact well) and be taken up efficiently into the neuron. When coming to the dopamine neuron, it is in a rigidified conformation which either 1) allows lower affinity binding, 2) still binds but fails to be transported into the neuron while DAT is changing conformation.
But when we consider the MDMA molecule, it can interact well with SERT by getting into a similar conformation as MDAI such that it is slightly depressed below the plane of the phenyl ring (like in MDAI, due to the indane moiety, the 2-amino group is depressed under the plane of the ring, similar to an axial substituent on cyclohexane), that is to say, the amino group in MDMA while bound to SERT may be in a gauche conformation, and the energy lost from the antiperiplanar conformation is less than the stabilisation energy from the molecule being bound to SERT. However, when MDMA comes to DAT, it will no longer have to be in a gauche conformation to bind, and just bind in its most stable conformation (antiperiplanar) where the N-methyl group is in the same plane as the ring. This makes sense as I'm guessing the natural substrate for DAT, dopamine, will be in this exact same antiperiplanar conformation when bound to DAT. Thus the MDMA binds effectively where as the MDAI, as the amino group is depressed below the ring and not it its plane, cannot bind effectively in its rigid conformation.
I might draw some pictures later to make my point clear.


I'm in doubt over whether the absence of neurotoxicity is related to MDAI's pharmacodynamic profile or if it's something to do with the actual molecule itself.
There is a lot of evidence that a lot of selective SRAs aren't neurotoxic due to the fact that not much DA release occurs and that not much DA can as a result be taken up into 5HT neuron and cause damage from there. For example, when a DRA and MDAI are co-administered, neurotoxicity is observed.
In terms of the molecule, the alpha methyl group is cyclised back onto the benzene ring. The structure is below for reference. I remember reading on wikipedia (can't seem to find on what page I read it, it might have been on the MDAI page itself but taken off for some reason) that because the alpha methyl is cyclised, there is no chance for methyl radicals to form unlike in MDMA, and that this plays a key in preventing cellular damage. I do not know much about radical chemistry at all, so if someone could evaluate these reasonings, that would be appreciated and spur interesting discussion.
Furthermore, I pondered over the fact that cyclising the molecule back to the ring could so drastically increase it's serotonin-dopamine selectivity (10:1 from wikipedia article). No new functional groups added, only a (small) change in conformation. Could it be that in fact because the molecule is rigidified and cannot rotate around the indane ring, it would always be in a decent conformation to interact with SERT (MDMA interacts better obviously but MDAI and its conformation definitely does interact well) and be taken up efficiently into the neuron. When coming to the dopamine neuron, it is in a rigidified conformation which either 1) allows lower affinity binding, 2) still binds but fails to be transported into the neuron while DAT is changing conformation.
But when we consider the MDMA molecule, it can interact well with SERT by getting into a similar conformation as MDAI such that it is slightly depressed below the plane of the phenyl ring (like in MDAI, due to the indane moiety, the 2-amino group is depressed under the plane of the ring, similar to an axial substituent on cyclohexane), that is to say, the amino group in MDMA while bound to SERT may be in a gauche conformation, and the energy lost from the antiperiplanar conformation is less than the stabilisation energy from the molecule being bound to SERT. However, when MDMA comes to DAT, it will no longer have to be in a gauche conformation to bind, and just bind in its most stable conformation (antiperiplanar) where the N-methyl group is in the same plane as the ring. This makes sense as I'm guessing the natural substrate for DAT, dopamine, will be in this exact same antiperiplanar conformation when bound to DAT. Thus the MDMA binds effectively where as the MDAI, as the amino group is depressed below the ring and not it its plane, cannot bind effectively in its rigid conformation.
I might draw some pictures later to make my point clear.
Last edited: