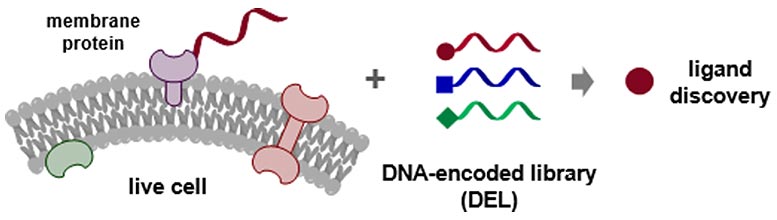
Chemists Develop a New Drug Discovery Strategy for “Undruggable” Targets
TOPICS:BiochemistryPharmaceuticalsThe University Of Hong Kong
By THE UNIVERSITY OF HONG KONG DECEMBER 29, 2020
DPAL
Graphic illustration of the work: DNA-programmed affinity labeling (DPAL) enables the direct screening of DNA-encoded chemical libraries (DELs) against membrane protein targets on live cells to create novel drug discovery opportunities. Credit: The University of Hong Kong
A research team led by Dr. Xiaoyu LI from the Research Division for Chemistry, Faculty of Science, in collaboration with Professor Yizhou LI from School of Pharmaceutical Sciences, Chongqing University and Professor Yan CAO from School of Pharmacy, Second Military Medical University in Shanghai has developed a new drug discovery method targeting membrane proteins on live cells.
Membrane proteins play important roles in biology, and many of them are high-value targets that are being intensively pursued in the pharmaceutical industry. The method developed by Dr. Li’s team provides an efficient way to discover novel ligands and inhibitors against membrane proteins, which remain largely intractable to traditional approaches. The development of the methodology and its applications are now published in Nature Chemistry, a prestigious chemistry journal by the Nature Publishing Group (NPG).
Cell Membrane DNA Tag
Artistic representation of the work: cell membrane is like a vast, complex, and unpredictable ocean. The membrane proteins are the rocks and islands in the ocean. Labelling the membrane protein target with a DNA tag is like having a lighthouse on the target protein to direct the specific screening of DNA-encoded chemical libraries for drug discovery. Credit: The University of Hong Kong
Background
Membrane proteins on the cell surface perform a myriad of biological functions that are vital to the survival of cells and organisms. Not surprisingly, numerous human diseases are associated with aberrant membrane protein functions. Indeed, membrane proteins account for over 60% of the targets of all FDA-approved small-molecule drugs. The G-protein coupled receptor (GPCR) superfamily alone, as the largest class of cell-surface receptors, are the targets of ~34% of all clinical drugs. However, despite the significance, drug discovery against membrane proteins is notoriously challenging, mainly due to the special property of their natural habitat: the cell membrane. Moreover, membrane proteins are also difficult to study in an isolated form, as they tend to lose essential cellular feature and may be deactivated. In fact, membrane proteins have long been considered as a type of “undruggable” targets in the pharmaceutical industry.
In recent years, DNA-encoded chemical library (DEL) has emerged and become a powerful drug screening technology. To simplify, we can use a book library as an example. In a library, each book is indexed with a catalog number and spatially encoded with a specific location on a bookshelf. Analogously, in a DEL, each chemical compound is attached with a unique DNA tag, which serves as the “catalog number” recording the structural information of the compound. With DNA encoding, all library compounds can be mixed and screened against the target simultaneously to discover the ones that can modulate the biological functions of the target, e.g. inhibiting the proteins that are aberrantly active in malignant cancers. DELs can contain astonishingly large numbers of test compounds (billions or even trillions), and DEL screening can be conducted in just a few hours in a regular chemistry lab. Today, DEL has been widely adopted by nearly all major pharmaceutical industry worldwide. However, DEL also had encountered significant difficulties in interrogating membrane proteins on live cells.
2 Key findings: Tracking and Boosting
There are two hurdles that the team has overcome to enable the application of DEL on live cells. First, cell surface is not a smooth convex shape like a balloon; it is extremely complex with hundreds of different biomolecules with a rugged topology; thus, locating the desired target on the cells surface is like finding a single tree in a thick tropical forest. The team has overcome this “target specificity” problem by using a method they previously developed: DNA-programmed affinity labeling (DPAL). This method utilizes a DNA-based probe system that can specifically deliver a DNA tag to the desired protein on live cells, and the DNA tag serves as a beacon to direct target-specific DEL screening. In other words, the team first installed a “tracker” on the target to achieve screening specificity.
The second challenge is target abundance. Typically, membrane proteins exist in nanomolar to low micromolar concentration, which is far below the high micromolar concentration needed to capture the tiny fraction of binders among billions of non-binders in a library. To solve this problem, the team employed a novel strategy by using complementary sequences in the DNA tag on the target protein and the actual library, so that the library can hybridize close to the target, thereby “boosting” the effective concentration of the target protein. In other words, the “tracker” can not only help the library locate the target, but also create an attractive force to concentrate the library around the target, not being distracted by the non-binding population.
In the publication, the team reports their detailed methodology development, and they also demonstrate the generality and performance of this method by screening a 30.42-million-compound library against folate receptor (FR), carbonic anhydrase 12 (CA-12), and epidermal growth factor receptor (EGFR) on live cells, all are important targets in anti-cancer drug discovery. This approach is expected to broadly applicable to many membrane proteins. For example, classical drug targets, such as GPCRs and ion channels, may be revisited in a live cell setting to identify new drug discovery opportunities by harnessing the power of DEL.
“We expect to the utility of this method is not limited to drug discovery, but also in academic research to explore challenging biological systems, such as oligomeric membrane protein complexes and cell-cell communications,” said Dr. Xiaoyu Li.
Co-corresponding author Professor Yizhou Li from Chongqing University said: “This method has the potential to facilitate drug discovery for membrane proteins with the power of large and complex chemical diversity from DNA-encoded chemical libraries.” Co-corresponding author Professor Yan Cao from Second Military Medical University in Shanghai added: “This technology is an effective tool for characterizing ligand-target interaction; it will cast new light on the development of high throughput screening methods, and thus facilitate the fishing of ligands targeting membrane proteins.”
Reference: “Selection of DNA-encoded chemical libraries against endogenous membrane proteins on live cells” by Yiran Huang, Ling Meng, Qigui Nie, Yu Zhou, Langdong Chen, Shilian Yang, Yi Man Eva Fung, Xiaomeng Li, Cen Huang, Yan Cao, Yizhou Li and Xiaoyu Li, 21 December 2020, Nature Chemistry.
DOI: 10.1038/s41557-020-00605-x
About the research team
The research was conducted by a team led by Dr. Xiaoyu Li from Research Division for Chemistry. Postdoctoral fellow Dr. Yiran Huang from Dr. Li’s group is the first author. Professor Yizhou Li from School of Pharmaceutical Sciences, Chongqing University and Professor Yan Cao from School of Pharmacy, Second Military Medical University in Shanghai are co-corresponding authors. Other HKU scientists in the Research Division for Chemistry contributing to the research include Miss Ling MENG, PhD student; Dr. Yu ZHOU, postdoctoral fellow; Dr. Yi Man Eva FUNG, Research Officer; Dr. Xiaomeng LI, postdoctoral fellow; and Mr. Cen HUANG, PhD student.
This work was supported by Research Grants Council of Hong Kong, Laboratory for Synthetic Chemistry and Chemical Biology of Health@InnoHK of Innovation and Technology Commission of Hong Kong, National Natural Science Foundation of China, and State Key Laboratory of Synthetic Chemistry at The University of Hong Kong. We thank the Centre for PanorOmic Sciences (CPOS) Genomics Core at HKU for analysis support.
About Dr Xiaoyu Li
Dr Xiaoyu Li is an Associate Professor of Research Division for Chemistry at The University of Hong Kong. His research interests lie in the fields of Chemical Biology, focusing on the development of new methods and enabling tools for both basic research and drug discovery. His research activities have been focusing on three areas: DNA-encoded chemical library (DEL) and its applications, protein labeling and profiling, and target identification and mechanism study of bioactive compounds.

 )
)