AlsoTapered
Bluelighter
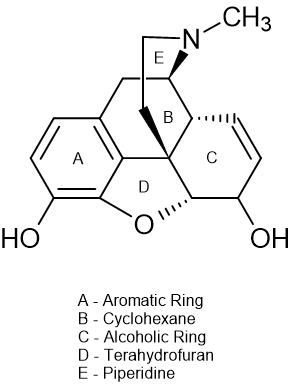

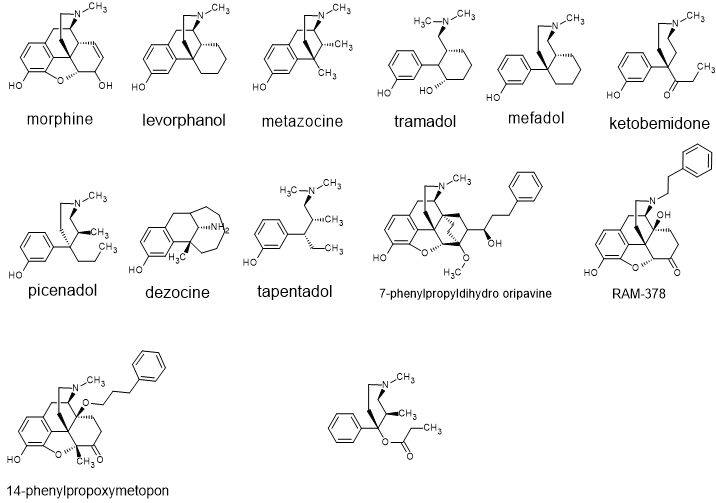
Let us begin with the prototype and most basic touch-stone for those developing novel opioids (definition - synthetic or semisynthetic derivatives of morphine).
I REALIZE that above giving names to the ring structures, this looks pretty dumb but trust me, I have a LOT to impart including stuff I have no answer to and so YOUR voices are important.
I've already built lesson II but I want to know people are OK with being presented with stuff they have no RATIONAL responses to because, as a medicinal chemist noted 'The QSAR of opioids it like 'Alice through the Looking Glass' and that's the guy who invented MXE put it, so he isn't an idiot... there are MANY things we just don't know.
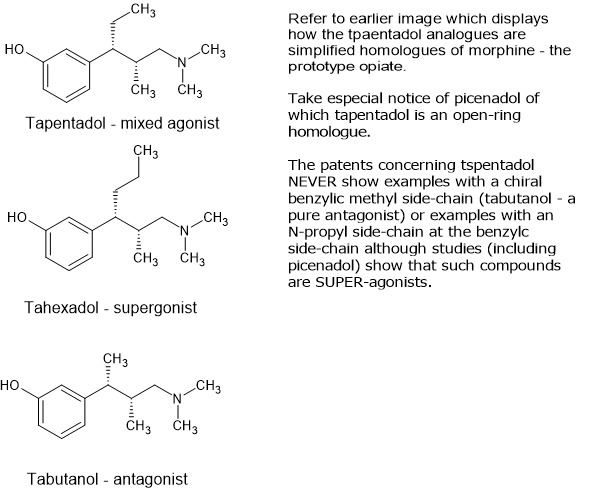
And a m-phenol function is the MOST tricky. If you didn't know, it isn't possible for an opiate ligand to be an ANTAGONIST without a m-OH,,, which is a simple rule but wasn't recognized until the late 1950s which is why 3-methyl ketobemidone (some 5.5 x morphine in potency) was never discovered at the time. Otto Eisleb (discoverer of MPPP, ketobemidone, prodine and so on) never spotted the fact. His job was to use the limited technology to produce the most potent phenylpiperidine opioid possible so MPPP, prodine and ketobemidine were his domain... but only 30 years later did someone work out that allylprodine, via careful testing. proved to be x23 M in potency. Adjust for MW and it's x8 M in potency.
I will attempt to provide facts and ask for people to suggest that which is not known. I mean, science not looking TOO hard. The KEY is, you know more than you think you do.
So we will explore ALL opioids. So step one is those strange names....
But let me tell you the absolute truth- ALL of you are more than able of understanding and designing novel opioids. It feels to me that it's almost the mystery of opioids that makes it feel as if non-chemists could NOT POSSIBLY understand. But I assure you, you are ALL capable of designing a novel and active opioid agonist.
Of course, the term opioid suggests semi-synthetic - But I hope to show how it's possible to learn the 'rules' of opioids and design your OWN opiate agonist.
I will not hide my goal. To make potent opioids so synthetically simple that EVERYONE can make them.
Last edited: