AlsoTapered
Bluelighter
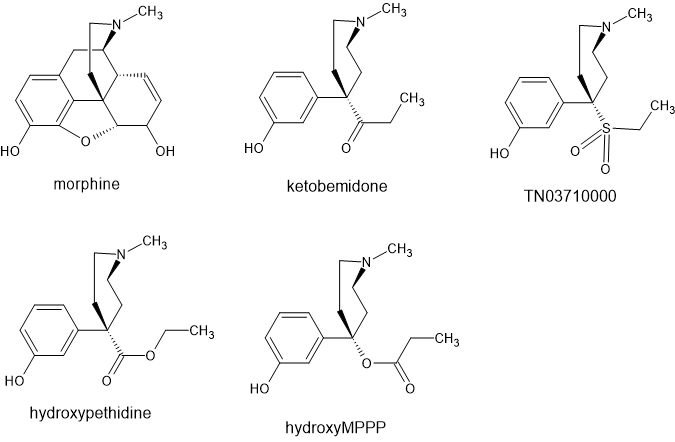
Otto Eisleb, the discover of pethidine went on to further develop the phenylpiperidine class of opioids. He developed MPPP, prodine and later ketobemidine.
Ketobemidine represents an almost unique compound having a meta phenolic group which substantially altered it's QSAR. Unlike pethidine derivatives, replacing the N-methyl with an N-phenylethyl resulted in ANTAGONIST activity. Unlike the prodine class, ring-substitution apparently resulted in ANTAGONIST activity.
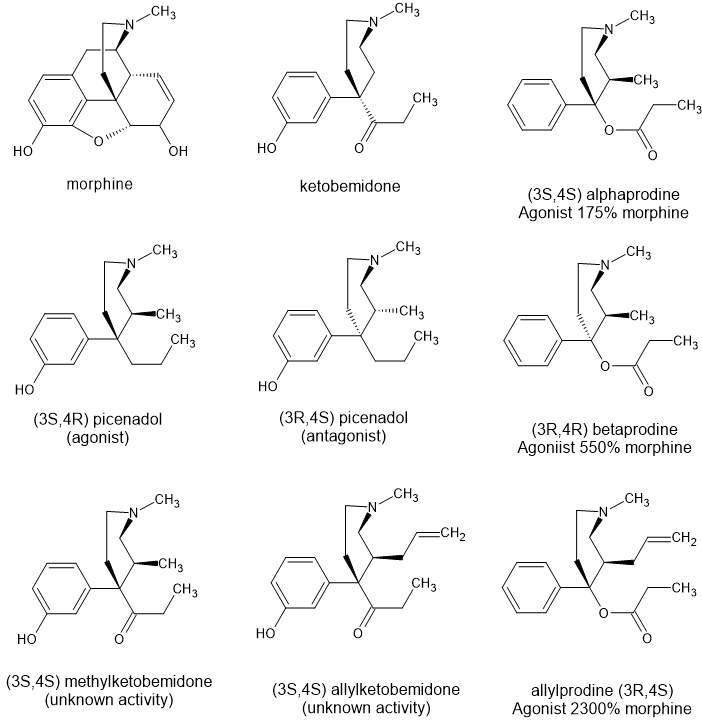
But here is the thing. While he was able to resolve the cis and trans enantiomers, he wasn't able to resolve the (R) and (S) isomers. Later a group at Eli Lilly discovered picenadol and discovered that one of the trans-pair was an agonist while the other was an antagonist. The mixture proved to provide analgesic activity and it was a least trialled briefly but proved to be of no clinical value.
So I've drawn the isomers of picenadol, of alpha and beta prodine and of allylprodine.
What I'm interested in knowing is if people think that the reason that Eisleb only obtained antagonist activity from 3-methyl ketobemidone was because the antagonist enantiomer (overlays beta prodine) is more potent than the agoist enantiomer (overlays alpha prodine).
Now, as far as I can tell nobody has been able to find the reason why ALL the enantiomers of prodine produce agonist activity while apparently, some enantiomers of picenadol (and so maybe ketobemidine) are agonists while others are antagonists.
Even the latest explainations are vague. It has been conjectured that phenolic and non-phenolic opioids bind at different sites within the receptor domain and the 3-substituuents 'increase receptor recognition' which is SO meaningless.
I said ketobemidine was almost unique because bemidone AKA hydroxypethidine (the phenolic homologue of pethidine/ and is listed as being an agonist with a potency some x1.5 that of pethidine itself. In fact, PubChem suggests that the phenolic homologue of MPPP was prepared but doesn't appear to be mentioned in the reference provided.
My last question is, if allylprodine displays much higher activity because it bind to 'an extra amino acid residue' then will allylketobemidone see a similar increase in potency?
It's a question worth asking because Danish and Swedish papers equate 25mg of ketobemidone with 60mg of morphine. Now ketobemidone has a significantly lower MW than morphine so that difference is even more pronounced. MPPP has about 70% the potency of morphine so that allyl increase potency by over 32 times (more when MW is adjusted) to infer that the appropriate allylketobemidone might be x82 morphine... but it's kind of fun to wonder.
after all, the law is VERY clear. Ketobemidone is controlled but no ketobemidone derivatives are covered because it was presumed that they would all be antagonists.
BTW two points. the ketone can be substituted with an ethylsulfonate in the same manner as methadone (whose bioisostere is IC-26) & some references suggest that the N-1-(furan-2-yl) homologue of ketobemidone has analgesic activity x25 that of morphine. Well, apparently that's due to kappa activity so is unlikely to be practical with the severe side-effects kappa ligands produce.
It is the way of things that in science, when a question is no longer of financial value, it gets very hard to get funding. But the fact that N-methyl is IT is very confusing. The phenolic version of pethidine and MPPP are both agonists BUT like ketobemidone, ONLY the N-METHYL!
Ketobemidine represents an almost unique compound having a meta phenolic group which substantially altered it's QSAR. Unlike pethidine derivatives, replacing the N-methyl with an N-phenylethyl resulted in ANTAGONIST activity. Unlike the prodine class, ring-substitution apparently resulted in ANTAGONIST activity.
But here is the thing. While he was able to resolve the cis and trans enantiomers, he wasn't able to resolve the (R) and (S) isomers. Later a group at Eli Lilly discovered picenadol and discovered that one of the trans-pair was an agonist while the other was an antagonist. The mixture proved to provide analgesic activity and it was a least trialled briefly but proved to be of no clinical value.
So I've drawn the isomers of picenadol, of alpha and beta prodine and of allylprodine.
What I'm interested in knowing is if people think that the reason that Eisleb only obtained antagonist activity from 3-methyl ketobemidone was because the antagonist enantiomer (overlays beta prodine) is more potent than the agoist enantiomer (overlays alpha prodine).
Now, as far as I can tell nobody has been able to find the reason why ALL the enantiomers of prodine produce agonist activity while apparently, some enantiomers of picenadol (and so maybe ketobemidine) are agonists while others are antagonists.
Even the latest explainations are vague. It has been conjectured that phenolic and non-phenolic opioids bind at different sites within the receptor domain and the 3-substituuents 'increase receptor recognition' which is SO meaningless.
I said ketobemidine was almost unique because bemidone AKA hydroxypethidine (the phenolic homologue of pethidine/ and is listed as being an agonist with a potency some x1.5 that of pethidine itself. In fact, PubChem suggests that the phenolic homologue of MPPP was prepared but doesn't appear to be mentioned in the reference provided.
My last question is, if allylprodine displays much higher activity because it bind to 'an extra amino acid residue' then will allylketobemidone see a similar increase in potency?
It's a question worth asking because Danish and Swedish papers equate 25mg of ketobemidone with 60mg of morphine. Now ketobemidone has a significantly lower MW than morphine so that difference is even more pronounced. MPPP has about 70% the potency of morphine so that allyl increase potency by over 32 times (more when MW is adjusted) to infer that the appropriate allylketobemidone might be x82 morphine... but it's kind of fun to wonder.
after all, the law is VERY clear. Ketobemidone is controlled but no ketobemidone derivatives are covered because it was presumed that they would all be antagonists.
BTW two points. the ketone can be substituted with an ethylsulfonate in the same manner as methadone (whose bioisostere is IC-26) & some references suggest that the N-1-(furan-2-yl) homologue of ketobemidone has analgesic activity x25 that of morphine. Well, apparently that's due to kappa activity so is unlikely to be practical with the severe side-effects kappa ligands produce.

It is the way of things that in science, when a question is no longer of financial value, it gets very hard to get funding. But the fact that N-methyl is IT is very confusing. The phenolic version of pethidine and MPPP are both agonists BUT like ketobemidone, ONLY the N-METHYL!