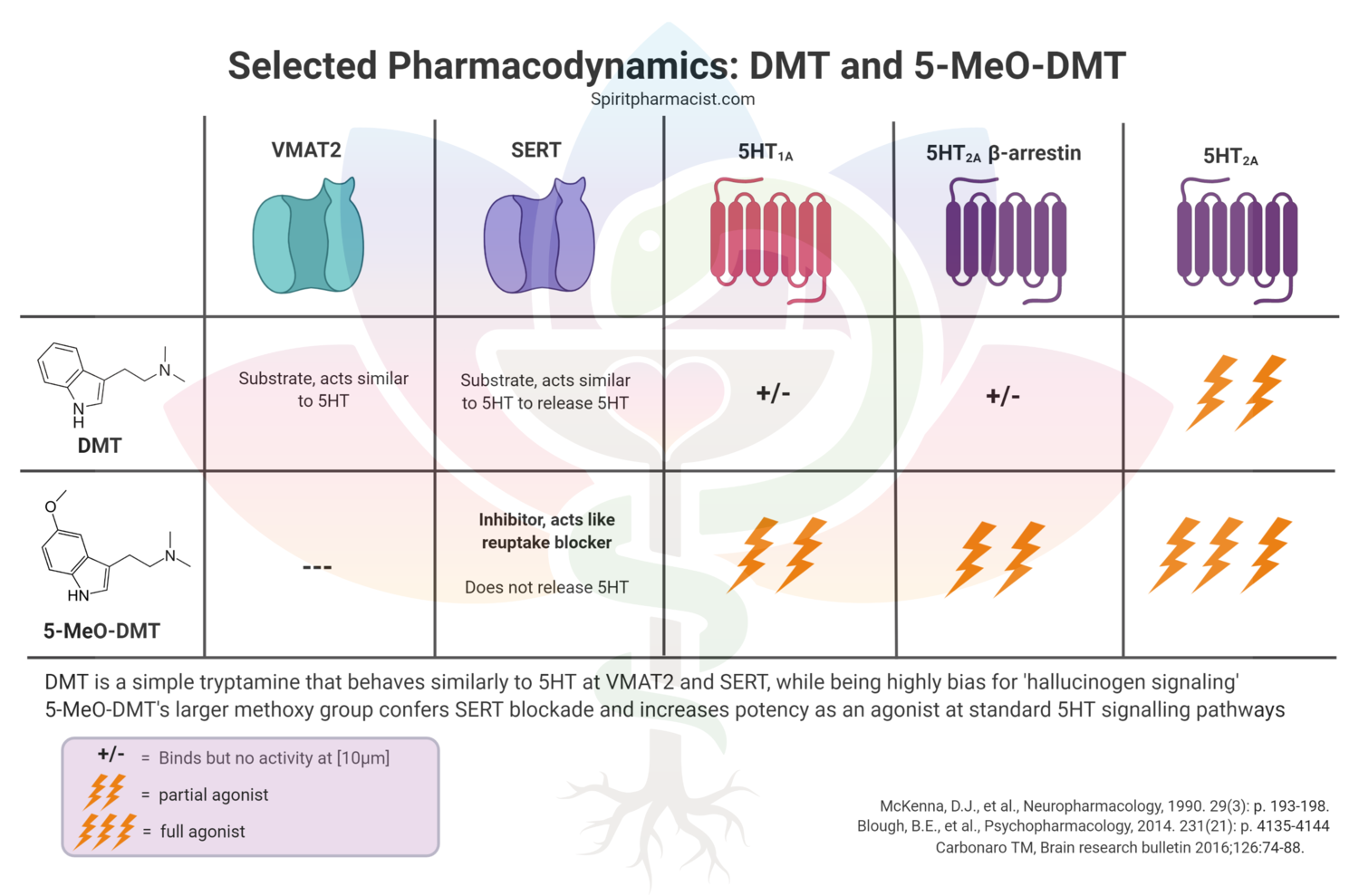
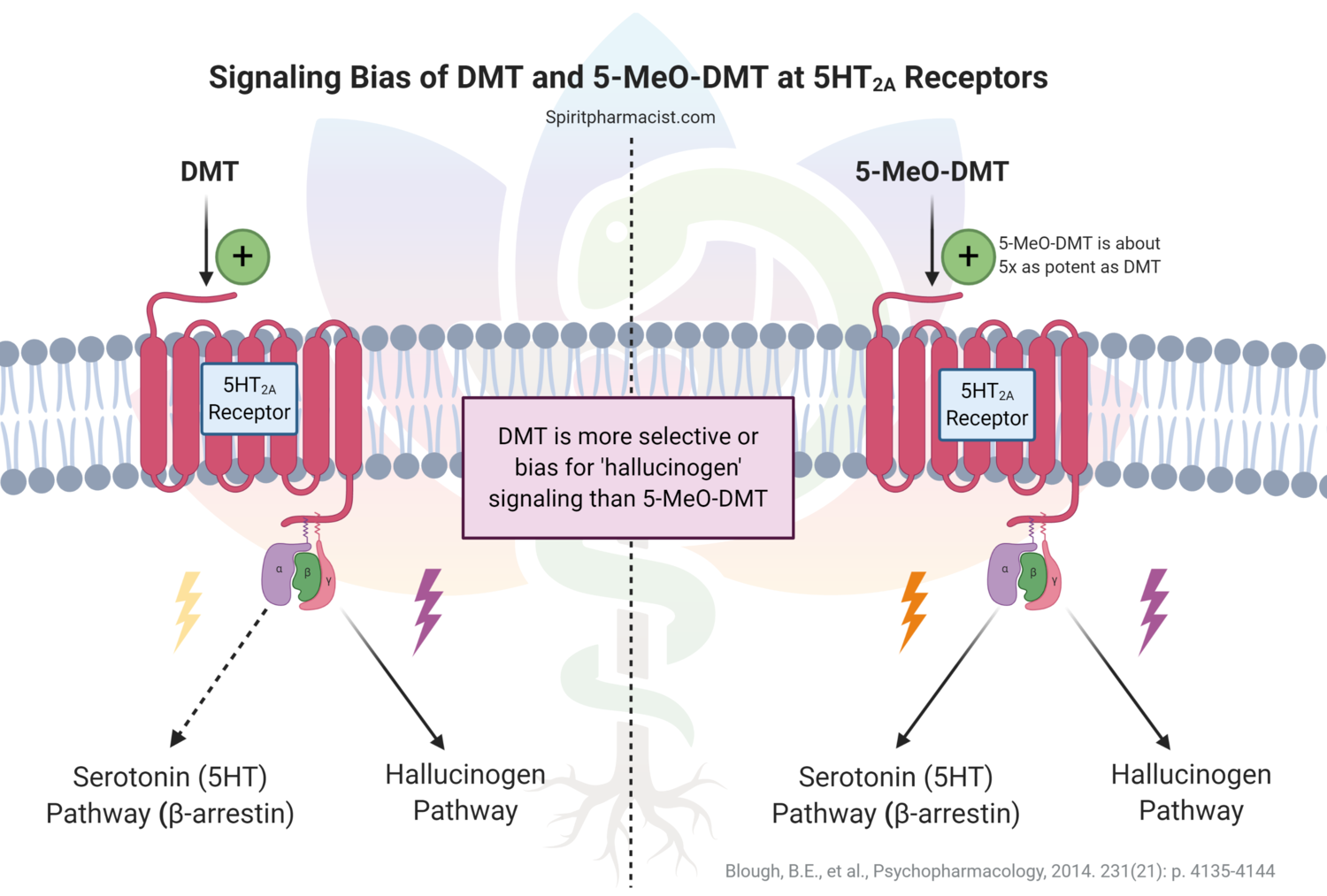
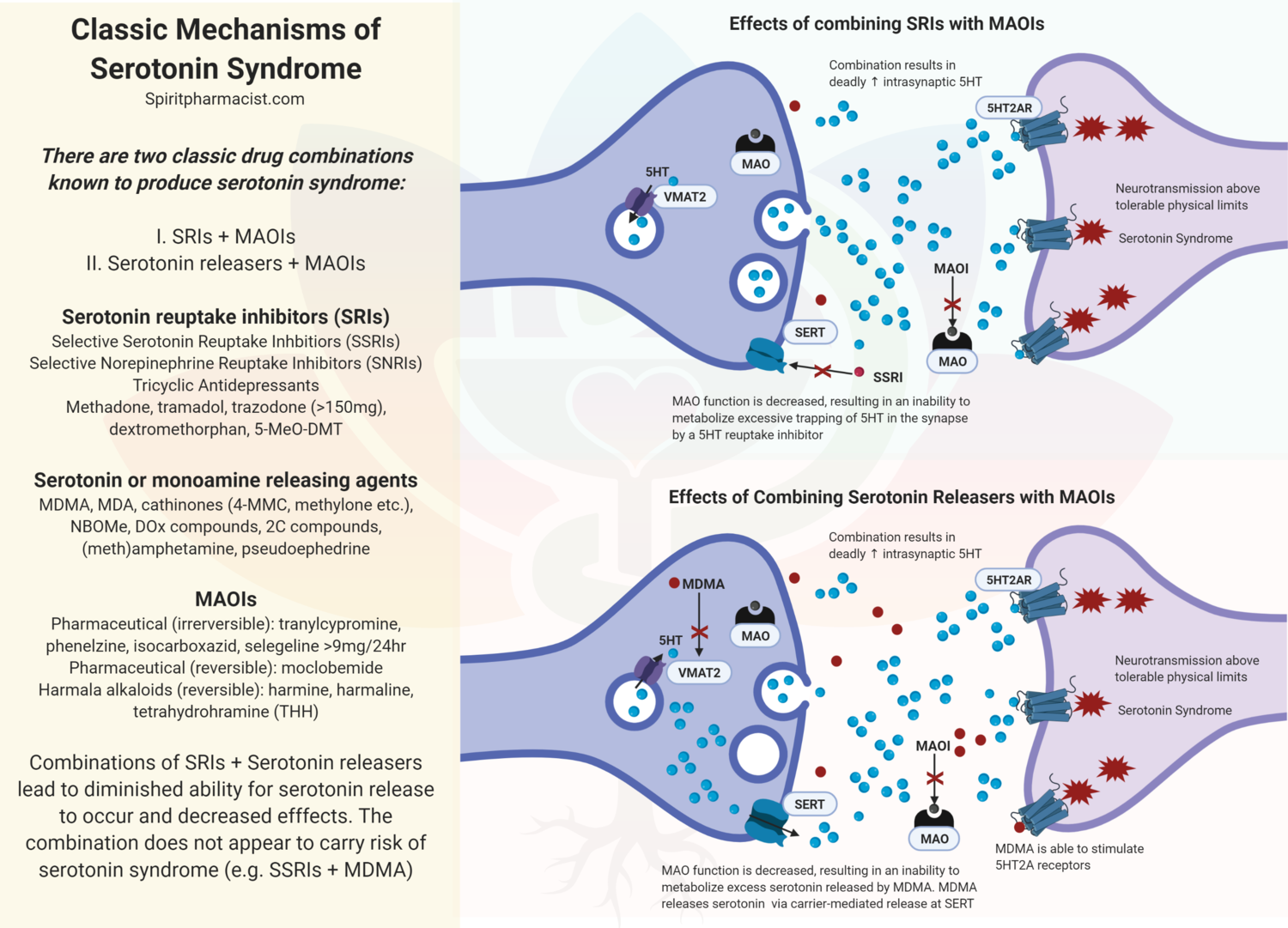
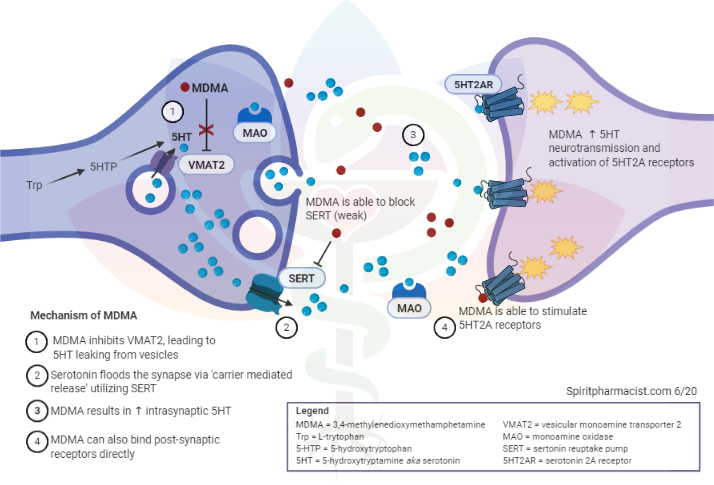
Assessing synthetic cannabinoids and cathinones in neuropsychiatric toxicological emergencies*
Sameer Hassamal, MD and Sunita Hassamal, MD | Psychiatric Times | 15 Jan 2021
Synthetic cannabinoids and cathinones are novel psychoactive substances (NPS) that have surreptitiously emerged on the market to mimic the effects of
established drugs. These designer substances are gaining popularity due to their widespread availability and safety misperceptions. Temporal trend epidemiological data analyzed by the Centers for Disease Control indicate that acute poisonings related to synthetic cannabinoids and cathinones are increasing in the
United States.
The rise in synthetic cathinone and cannabinoid use among patients susceptible to adverse neuropsychiatric reactions has been linked to a wide range of unpredictable effects, including
agitation, psychosis, and delirium.3 Emergency department (ED) psychiatrists are on the frontlines of care, and must provide the initial management and stabilization of the complex and challenging toxic effects of these drugs. Prompt recognition and management is essential to reduce morbidity and mortality.

Table 1. Common Adverse Neuropsychiatric Effects of Synthetic Cannabinoid and Cathinone Intoxication.
Synthetic cannabinoids
Colloquially known as
spice, these designer synthetic cannabinoid receptor agonists have potent intrinsic activity at the cannabinoid (CB) 1 and 2 receptors. The compounds were originally developed to study the endocannabinoid system, but they emerged as a drug of abuse in 2008.5 Initially designed to mimic the effects of delta 9-tetrahydrocannabidiol, synthetic cannabinoids are 2 to 100 times more potent than marijuana at the CB1 and CB2 receptor, resulting in a wide range of adverse neuropsychiatric symptoms.
(Table 1)
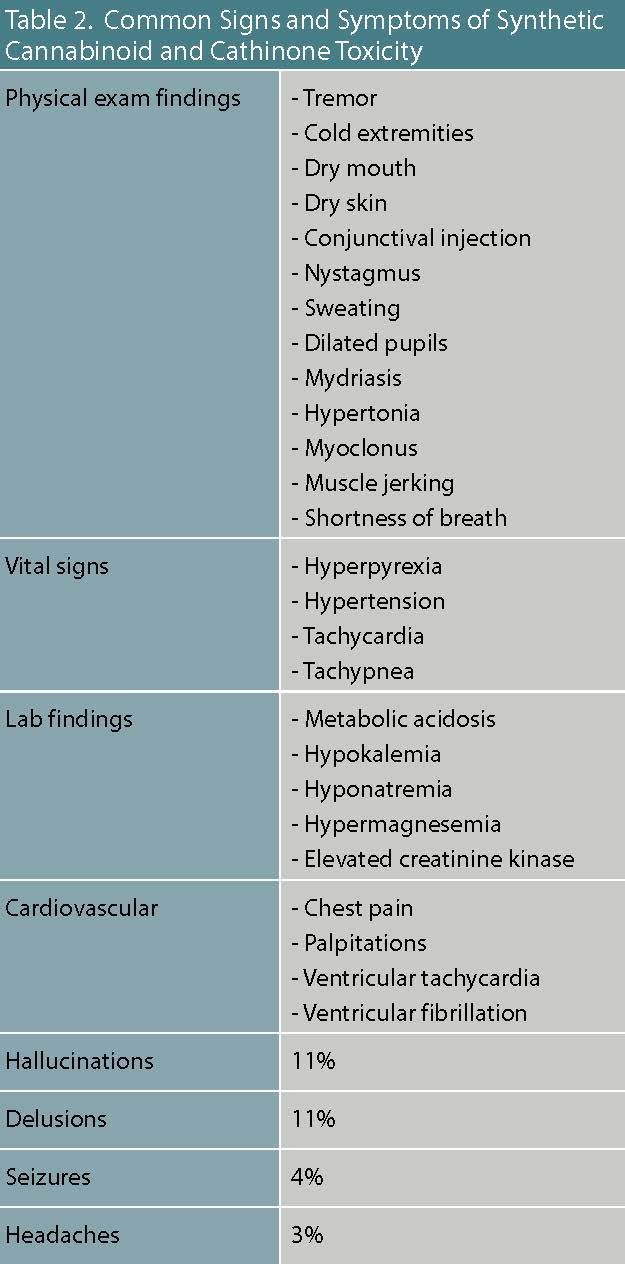
Furthermore, synthetic cannabinoids are often adulterated with additional ingredients to either enhance or attenuate the drug high, resulting in clinically unpredictable toxicological profiles. As a consequence, there is no specific toxidrome associated with synthetic cannabinoid intoxication, and patients will typically present with a constellation of sympathomimetic, anticholinergic, and marijuana toxic effects. (
Table 2). The most common sympathetic nervous system cardiovascular effects are tachycardia (46 percent) and hypertension (21 percent). The anticholinergic effects closely overlap with signs and symptoms of a sympathomimetic toxidrome.
Distinguishing symptoms specific to anticholinergic toxicity include: dry skin, cotton mouth, absent bowel sounds, blurred vision, and urinary retention. Moreover, patients may additionally present with marijuana intoxication symptoms (ataxia, delayed reaction time, and
conjunctival injection). Laboratory studies are indicated in cases of severe intoxication with agitation or seizures due to the resulting life-threatening metabolic derangements.

Table 2. Common Signs and Symptoms of Synthetic Cannabinoid and Cathinone Toxicity
Rates of synthetic cannabinoid use are increasing in the United States, with the lifetime prevalence estimated to be between 0.2% and 4%. Moreover, between January 2016 and September 2019, ED visits related to synthetic cannabinoid toxicity significantly increased in the country’s Midwestern and Northeastern regions. Overall, synthetic cannabinoids harms are increasing, and psychiatrists need to be cognizant about the clinical presentations and evidence based treatments.
Synthetic cathinones
Colloquially known as
bath salts, these agents are synthetic psychostimulants structurally related to cathinone, a monoamine alkaloid found in the Khat plant. As substituted phenethylamines, synthetic cathinones are structurally and pharmacologically similar to amphetamine and 3,4-methylenedioxymethamphetamine (MDMA). These agents are commonly abused for their euphoric, empathogenic, and stimulating effects. The most commonly abused synthetic cathinones are 4-methylmethcathinone (mephedrone), 3,4-methylenedioxy-N-methylcathinone (methylone), and 4-methylenedioxypyrovalerone (MDPV). Synthetic cathinones produce major effects on the Central Nervous System (CNS), and are linked to adverse neuropsychiatric complications (
Table 1). Synthetic cathinones stimulate the release of dopamine and inhibit the reuptake of epinephrine, norepinephrine, and serotonin inducing a broad range of sympathomimetic, serotonergic, and hallucinogenic adverse toxidrome effects (
Table 2). The most frequent sympathetic nervous system cardiovascular effects are tachycardia (46 percent) and hypertension (21 percent). The hallucinogenic effects (perceptual distortions, depersonalization, synesthesia) can be highly distressing, which often prompts emergency psychiatric care. Ancillary lab studies should be ordered in cases of severe agitation because of the increased risk for acute kidney injury, rhabdomyolysis, lactic acidosis, stroke, and intracranial hemorrhage.
Self-reported prevalence of synthetic cathinone use is an estimated 0.05%. Current estimates are imprecise, however, as established surveys and standard immunoassay testing are not sensitive for detecting cathinones. The most recent data from the Drug Abuse Warning Network report indicates 22,904 ED visits were related to synthetic cathinone toxicity in 2014. The frequency of ED utilization is likely increasing, as synthetic cathinones are steadily becoming more popular despite the potential harms.
General management principles
The diagnosis of intoxication is based on a history of exposure and presence of characteristic signs and symptoms. Patients may unknowingly or inadvertently administer synthetic cannabinoids and cathinones, as these compounds are oftentimes adulterated with other illicit drugs such as MDMA. Although there is no specific toxidrome, patients will typically present with signs and symptoms of adrenergic neurohumoral activation. The neurotoxic symptoms (including aggression, confusion, and psychosis) may overlap with a primary mental illness, and psychiatrists must rely on the clinical history and physical exam findings to accurately diagnose toxicity. Urine immunoassay testing is the most common method to screen for substance use. In the setting of acute intoxication, however, urine immunoassay testing has a low positive predictive value for synthetic cannabinoids and cathinones. While gas chromatography/mass spectrometry (GC/MS) is considered the gold standard for detecting substances, the molecular geometry of synthetic cannabinoids and cathinones is everchanging, making it increasingly difficult to directly identify these compounds. Moreover, GC/MS is not readily available in all emergency hospitals, and the results may not return in a timely manner to aid in the diagnosis process. An atypical onset of psychosis and agitation alongside hyper-sympathetic activation and a mixed toxidrome is strongly suggestive of intoxication.
Because of the variability in clinical presentations and lack of diagnostic testing, psychiatrists should strongly consider the possibility of intoxication in any patient with no known psychiatric history, a negative urine drug screen, and no known contributing medical mimickers (presenting with a mixed toxidrome). Patients will not necessarily fit neatly into a particular toxidrome, but rather present with overlapping signs and symptoms from multiple toxidrome classes. Consequently, differentiating synthetic cannabinoid and synthetic cathinone toxicity can be challenging. Regardless, supportive care and symptom management is the mainstay of the treatment. Benzodiazepines are considered the first line of treatment to reduce the sympathomimetic neural stimulation (including hypertension, agitation, and seizures).
Agitation
Agitation is the most common neuropsychiatric complication associated with synthetic cannabinoid and cathinone ingestion. Katz and colleagues reported that 40% of intoxicated patients evaluated in an ED will experience severe agitation. The spectrum of psychomotor agitation can be objectively measured and identified with a standardized rating scale such as the Behavioral Activity Rating Scale (BARS). For patients with mild-moderate agitation, the patient may initially be calmed with verbal de-escalation techniques, and environmental modification. For patients with moderate-severe agitation, benzodiazepines remain the first line treatment for reducing moderate to severe agitation. Among the benzodiazepines, midazolam and lorazepam are considered the initial drugs of choice for decreasing agitation because of their rapid onset of action, short half-life, and parenteral route of administration availability. Antipsychotics are commonly administered with benzodiazepines to manage the psychotomimetic effects. However, risks associated with administering antipsychotics in acute poisonings settings must be carefully considered. Specifically, antipsychotics: may increase the risk of seizure induction; have anticholinergic effects; impair thermoregulation mechanisms; and prolong the QTc interval. Serotonergic sedatives such as fentanyl should be avoided because of the increased risk of serotonin toxicity associated with synthetic cannabinoids and cathinones.
Excited delirium
A subset of patients will develop excited delirium following ingestion of psychostimulants. A systematic review reported that 25% to 94% of patients diagnosed with excited delirium were recently exposed to synthetic cannabinoids and/or cathinones. The American College of Emergency Physicians has provided guidance on evaluating excited delirium: a minimum of 6 out of 10 clinical criteria are required to make a diagnosis. If undertreated, the case-fatality rate is estimated to be 20%, partly due to catecholaminergic cardio-respiratory failure. The cornerstone of treatment is rapid sedation to reduce the sympathomimetic outflow and facilitate lifesaving treatments.
To date, no large scale randomized controlled trials have evaluated pharmacological treatments for excited delirium. Nonetheless standard of care is to administer benzodiazepines and antipsychotics to decrease psychomotor agitation. In the emergency setting, IM ketamine (4-5 mg/kg) is increasingly used for sedation of agitated patients with excited delirium. Initial studies in the pre-hospital setting (emergency medical services) reported that intubation rates following ketamine administration were as high as 60%. Emerging literature supports that IM ketamine administration in the hospital setting (emergency department) is a safe and effective treatment for excited delirium while not significantly affecting the cardiopulmonary system. Mankowitz and colleagues reported that more patients required intubation after ketamine administration in the pre-hospital setting compared to the hospital setting. Prospective studies conducted in the emergency department have demonstrated that there is no significant difference in incidence of complications between IM ketamine and IM haloperidol. The side effects of single doses of IM ketamine for agitation are generally transient and self-limiting: emesis, hypersalivation, recovery agitation (also known as emergence reaction), hypertension, and tachycardia. Moreover, ketamine has other potential benefits, including a faster onset to sedation compared with benzodiazepines and antipsychotics. There are observational reports of ketamine exacerbating psychosis in susceptible patients. Larger studies, however, have concluded that acute- and short-term administration of ketamine for agitation does not increase psychiatric inpatient admissions or ED psychiatric evaluations. Physical restraints should be used as a last resort, primarily as a bridge to pharmacologic treatment, as prolonged use of physical restraints is likely to worsen delirium and rhabdomyolysis, leading to renal failure, electrolyte abnormalities, and arrhythmias.
Psychosis
Synthetic cannabinoids have been linked to psychotic reactions, such as paranoia, catatonia, dissociation, auditory, and/or visual hallucinations. Longitudinal studies have reported that consumption of potent cannabinoids quintuples the risk of a psychotic episode. The psychotomimetic effects are usually transient and resolve within 5 to 8 days; however, up to one-third of patients will transition to schizophrenia. Although there is no established antidote, preliminary reports suggest that cannabinol, an allosteric modulator at the CB1 receptor, may reverse the neurotoxic effects.
Similarly, synthetic cathinones have pro-psychotic effects, potentially triggering a psychotic state in vulnerable groups of patients. An observational study reported that synthetic stimulants were implicated in approximately 25% of ED visits involving drug-related psychosis. Specifically, mephedrone was the most common synthetic cathinone involved in stimulant psychosis, accounting for approximately 20% of stimulant related psychotic cases. Synthetic cathinone induced psychosis is typically brief and resolves within days to weeks; however, 25% of patients will transition to a schizophrenia spectrum disorder.
The psychotomimetic symptoms can be controlled with first- and second-generation antipsychotic medications with careful consideration of side effect profiles. Among the first-generation antipsychotics, the butyrophenone neuroleptics IM haloperidol and IM droperidol are reasonable pharmacotherapeutic options. In comparison to IM haloperidol, IM droperidol has a faster onset of action, rapidly preventing and treating emergent psychotic agitation within 5 to 10 minutes of intramuscular administration.36 Some expert panels have recommended avoiding butyrophenone neuroleptics in cases of substance-induced psychosis due to the pro-convulsant properties, cardiotoxic effects, and extrapyramidal symptoms. Among the second-generation antipsychotics, p.o. olanzapine has a track record of treating NPS-induced psychosis, and pro-drug websites have touted olanzapine as the ideal medication to terminate so-called “bad trips.” In spite of its efficacy, olanzapine has anticholinergic and central thermoregulatory effects impairing core temperature regulation. In theory, p.o. or IM aripiprazole and ziprasidone are probably the safest antipsychotics to administer because of the relatively favorable safety profile, and negligible anticholinergic and seizurogenic effects.
Despite the infamous concerns about
QTc prolongation, epidemiological studies do not support a correlation between ziprasidone and torsade de pointes. In many instances, detoxification with benzodiazepines and rest will often quell the substance-induced symptoms without the added need for antipsychotics. Given the risks associated with neuroleptics, especially in antipsychotic naïve patients, it may be best to consider a benzodiazepine as the first line agent to reduce the psychotic agitation. Antipsychotics can be initiated if the psychosis continues to persist after the detoxification period, or if the patient has a underlying primary psychotic illness.
Concluding summary
Synthetic cannabinoid and cathinone harms are increasing, and psychiatrists need to be cognizant of the clinical presentations and evidence-based treatments. Synthetic cannabinoids and cathinones have pro-psychotic and agitative effects, and the toxic reactions can mimic a primary mental illness. There is no definitive diagnostic test to differentiate primary versus secondary drug induced mental illness, and ED psychiatrists must rely on the history and physical examination. In a subset of cases, the overstimulation of catecholamines may result in an excited delirium. Benzodiazepines are considered the first line treatment to reduce the sympathomimetic neural stimulation.
*From the article here :
Be on the lookout for these novel psychoactive substances.
www.psychiatrictimes.com


 www.news-medical.net
www.news-medical.net