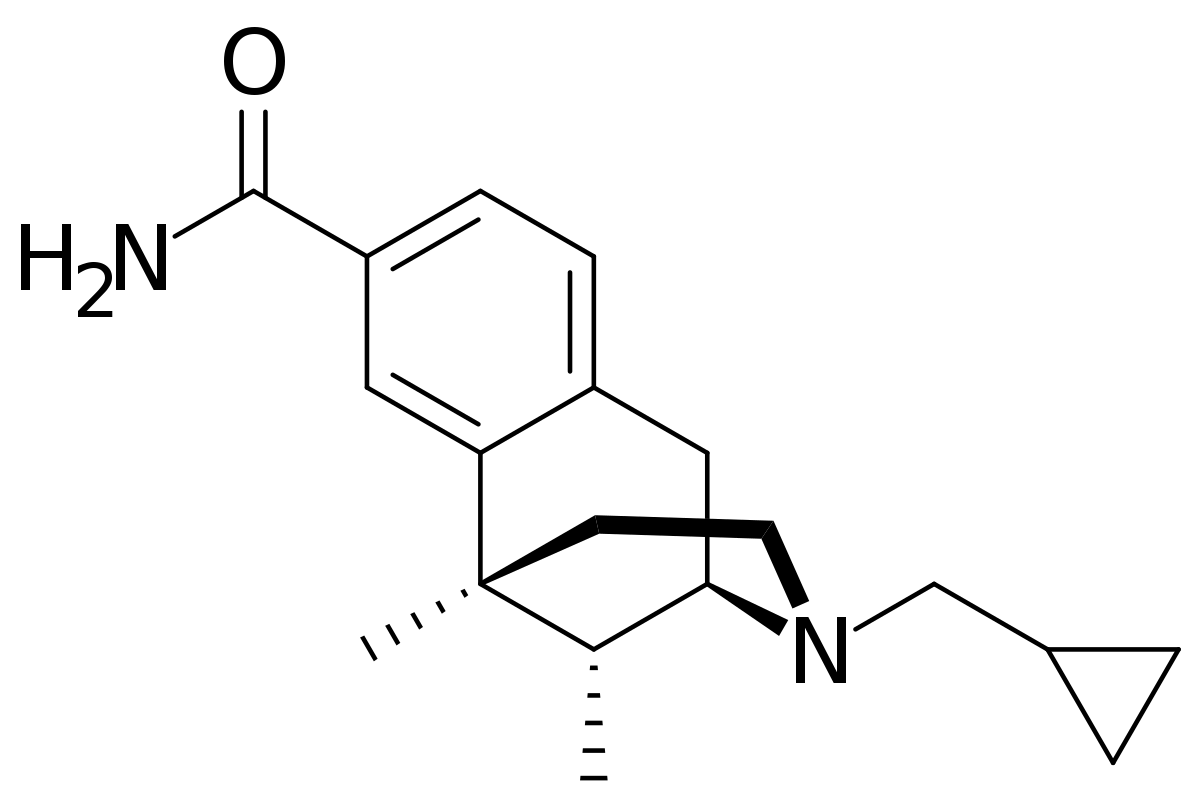
A good question might be - how would one modify levorphanol to increase it's duration of action? To do that you need to know the metabolism. Well I will tell you, N-demethylation (and norlevorphanol is considered active in it's own right) and by ester formation at the phenolic 3 position.... so one would need to explore what else could go at 3.
The answer is again, that place I say we do not accept references from BUT use references IN:

en.wikipedia.org
Swapping the phenol for a carboxamide slightly reduced potency but read the references and the T1/2 increases by a factor of 8.
Now that would see a huge accumulation of the nor metabolite and frankly an analgesic that lasts for days is generally a bad plan, but it's EXACTLY the sort of question someone might ask.. so I would HAVE all of those references ready, a rationale of why it WOULD would similarly in levorphanol and even a sketch of the synthesis (which is actually just 2 steps).
But here we post facts or ask questions. EVERYONE is welcome to ask questions... but 'I might have read it wrong' I am afraid, isn't likely to win friends or influence people.
In short - it's a LOT of work. If you come up with 1 novel thing a week you will be doing well. I have 34 years of experience to rest on so maybe I don't recall WHICH paper, but I know it was in a paper I read and I will spend days (if needed) finding that paper.
I don't mean to be hard on you, but we have 1 muppet who if I had any control wouldn't be able to post here. As it is they have their own thread and nobody else even bothers to look. I advise you to avoid them like the plague. They WANT to convince someone, ANYONE that they are a genius.
I most certainly am not, but I do work hard. Their is no substitute for that.