-
Neuroscience & Pharmacology Discussion Welcome Guest
Posting Rules Bluelight Rules Recent Journal Articles Chemistry Mega-Thread FREE Chemistry Databases! Self-Education Guide -
N&PD Moderators: Skorpio
You are using an out of date browser. It may not display this or other websites correctly.
You should upgrade or use an alternative browser.I Like to Draw Pictures of Random Molecules
- Thread starter nuke
- Start date
- Status
- Not open for further replies.




pharmakos
Bluelighter
Isnt that an adamant-1-yl?
yup.
in confirming my "yup" i realized something i never saw in the adamantyl group before... its essentially a polygon made up of 4 hexagons. =p or 4 overlapping cyclohexyl rings, if you will. certainly wouldn't look like a regular polygon in reality, but yeah.APINACA is an example of a drug containing the moiety, also amantadine and memantine..
if you always wanted to feel adamantyls in your body (I don't believe you find them in nature, apart from adamantane in oil) Hehe they classify it a tricycloalkyl... the fourth one is implied ;p you can't count it without "breaking the fourth wall".
(I don't believe you find them in nature, apart from adamantane in oil) Hehe they classify it a tricycloalkyl... the fourth one is implied ;p you can't count it without "breaking the fourth wall".
Roi, is that a secondary amine? ^ I'm curious..@Solipsis
That roi's opioid's amine has a phenethyl attached to it, so tertiary.
@pharmacos
lol that's the "beauty" of adamantyl, or a diamond. If you interest in this kind of shape try googling nanodiamond (or diamondoid) structure or so,
they are like adamantyl bulked with each other side by sides; in the same way of forming diamond structure.
Yet they are still organic molecule (even with vapor pressure; sublimable slowly) and has hydrogens around in the outer.
DrugDesigner
Greenlighter
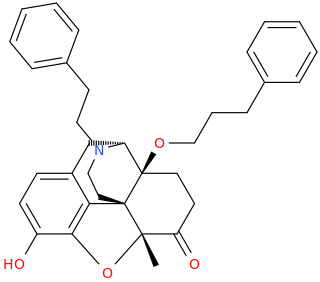
Would the molecule I designed be Psychoactive or psychedelic at all? I started with the classic bromo-hemiFLY structure, and then added a D-methamphetamine structure to it sharing the same benzene ring, I also added the structure for DMT on the bottom sharing the same benzene ring as the other two. Basically We have three different compounds that are all psychoactive on their own, and then combining all three into one big compound with all three compounds sharing the same singular benzene ring at the center.
I am really interested in designing psychoactive molecules especially psychedelic ones. Very interested to learn and understand pharmacology and the chemistry behind this. Is there any way I could virtually test this novel compound for possible psychoactivity on my computer? or like a program that simulates and predicts the psychoactivity or pharmacological action of a novel compound? I would love to be able to somehow test novel compounds for psychoactivity and binding affinity and such without having to do it in real life - that would be basically impossible. I am already using a program called ToxTree to test for toxicity, but no matter what even for water it always says that every compound I enter in is extremely toxic. Are there any other GUI programs for mac that accurately test for toxicity?
TL;DR: designed this compound, wondering if it would be psychoactive/psychedelic in effects. Looking for any way to virtually test or simulate the pharmacological action of compounds on the computer or online
[/URL][/IMG] Last edited:@Solipsis
Last edited:@Solipsis
That roi's opioid's amine has a phenethyl attached to it, so tertiary.
Oh yea, thought it was on the morphinan, it wasn't clear - though that clearly would have been too unusual a place for a substitution, esp one like that, wasnt using my head.
Would the molecule I designed be Psychoactive or psychedelic at all? I started with the classic bromo-hemiFLY structure, and then added a D-methamphetamine structure to it sharing the same benzene ring, I also added the structure for DMT on the bottom sharing the same benzene ring as the other two. Basically We have three different compounds that are all psychoactive on their own, and then combining all three into one big compound with all three compounds sharing the same singular benzene ring at the center.
I am really interested in designing psychoactive molecules especially psychedelic ones. Very interested to learn and understand pharmacology and the chemistry behind this. Is there any way I could virtually test this novel compound for possible psychoactivity on my computer? or like a program that simulates and predicts the psychoactivity or pharmacological action of a novel compound? I would love to be able to somehow test novel compounds for psychoactivity and binding affinity and such without having to do it in real life - that would be basically impossible. I am already using a program called ToxTree to test for toxicity, but no matter what even for water it always says that every compound I enter in is extremely toxic. Are there any other GUI programs for mac that accurately test for toxicity?
TL;DR: designed this compound, wondering if it would be psychoactive/psychedelic in effects. Looking for any way to virtually test or simulate the pharmacological action of compounds on the computer or online
[/URL][/IMG]
http://www.bluelight.org/vb/threads...-molecules?p=13546642&viewfull=1#post13546642
Your molecule combines phenethylamine and tryptamine backbones, but there are a few problems:
- you have two separate ethylamine / propanamine backbones there on the right side
- a hybrid is already problematic because the substitutions are not mutually compatible, and even less when they are not overlaid properly which is what happened here and what causes the double sidechain. If you check my link, the top two show ways there is I think a better matching overlay, hybridizing the structures.
- like I said: some things like the bromo from the PEA is probably not well tolerated on that position on the tryptamine - you are trying to put a number of features in there that really push the limits - it's best to first keep it as simple as possible. Simpler than mine as well, cause I was just playing around really. But to summarize, I'd remove that methaminopropyl group on the top right and the bromo to get a constrained 5-MeO-DMT. Regarding methylenedioxy PEAs with the MDO at a different place than the 3,4 in MDMA, they should be present as entries in PiKHAL.
- I think hybrids should be designed very carefully, often the difference (between say PEA and tryptamine) means some sort of different geometry with regard to the receptor, so if you modify the backbone so heavily, it may be moved a bit, and suddenly you get very different tolerance for groups that were previously very effective. Inconsistencies like that can cause a hybrid to try and satisfy two different models, but neither really applies anymore so it fails at both.
Occasionally hybrids to turn out to be possible, which is IMO always pretty awesome.
Also
http://www.bluelight.org/vb/threads...-molecules?p=13637699&viewfull=1#post13637699
Testing the structure requires software for computational binding studies..
https://en.wikipedia.org/wiki/Molecular_design_software
https://en.wikipedia.org/wiki/Quantitative_structure–activity_relationship
If designing more simple analogues, you can try overlaying models that calculate similarity of the physical properties, some things can be inferred from that I think.
But ideally wait for the professionals to "cheme" inLast edited:
DrugDesigner
Greenlighter
Thanks for the input @solipsis
It seems that I am on a kick with -FLY derivatives. They interest me a lot. I was wondering if any of the following -FLY derivative would be in theory psychoactive at all.
Thanks!
[/URL][/IMG] Last edited:Bromo-Dragonfly is actually more like a trivial name for DOB-Dragonfly ("Bromo" specifies nothing about the sidechain), so the upper compound is DOB-FLY.
Last edited:Bromo-Dragonfly is actually more like a trivial name for DOB-Dragonfly ("Bromo" specifies nothing about the sidechain), so the upper compound is DOB-FLY.
As your 'bromo-butterfly' is similarly a suggested trivial name for DOB-butterfly, the bottom one cannot be TFM-butterfly but must be DOTFM-butterfly because of the alpha-methyl.
I'm not certain if you can use the phrase 'fully aromatic' like that, maybe you can - but the stingray compound is not entirely planar (flat) because the methoxy part of the 'wings' will affect the angles because of the hybridization of the type of bonds oxygen makes. Call it unsaturated instead.")
2C-B-FLY is not far off in dosage and duration to the parent compound 2C-B, only it has constrained methoxies which may help selectivity of action like LSZ compared to LSD.
DOB-FLY would really be expected to be similar to DOB in dosage and duration but again with slightly different or more selective effect, as the conformation may not satisfy all receptor binding energies quite as easy as a floppy methoxy. It would be interesting to see how stimulating it feels, as apparently despite being amphetamines DOX don't really behave like amphetamines at all I think, well according to @Ebola? . More selective effect could fix the stimulation despite still technically being an amphetamine. Or for all we know it could be just different or a bit worse.
All kinds of combinations of structure like Butter- Hemi- etc have been proposed, but afaik they are not that easy to synthesize and are largely unexplored. It's really excellent that 2C-B-FLY is on the market, but it may already be pushing the market 'demands'. It would be great to see another niche constrained PEA, but I think it will only happen if there is a particularly excellent idea that is experimentally sampled and found to be a really special gem.Last edited:
belligerent drunk
Bluelight Crew
- Joined
- Sep 16, 2015
- Messages
- 3,482
Functional NMDA antagonists like the ACA's are modelled on NMDA and electronically mimic the carbonic acid functions of NMDA using aromatic groups loosely. While they are not exactly the heavily polar types that appear to often act as glutamate or glycine site ligands, they still carry the NMDA signature, pretty unmistakably. I personally doubt that you can just substitute the phenyl ring which is already a 'modification' into a bioisostere (i.e. broadly resembling) of a modification - that is 2 degrees removed. Could you just swap the cyclohexane in ACA's for another phenyl? Again, I doubt it since it may heavily undermine the tolerance for changes in the electronic configuration.
It seems to me that it would be so offensive to the NMDA pharmacophore that there is either a loss of potency or overall function to be expected from it.
Are you sure about that? I know that it's a pretty popular thought, but seems rather bullshit to me.

NMDA's both carboxyls would be deprotonated and would have a negative charge, which is significantly different from, in this example, an aromatic methoxy- and regular keto groups. And also, what's with the rest of the aliphatic and aromatic bulk? To me, it doesn't look like an NMDA bioisostere at all.
ACAs bind to the PCP site, don't they? PCP itself doesn't have oxygens at all, so it only strengthens the point that they don't mimic NMDA's electron structure at all, even if their structures on paper may "look NMDA-like".Last edited:
pharmakos
Bluelighter
was covered a few pages back in this thread IIRC... i believe glutamate and MXE bind to different sites on the NMDA receptor, so any structural similarity is merely coincidental
Nagelfar
Bluelight Crew
Isnt that an adamant-1-yl?
Are those actually two differing conformations of the ring that exist in physics? What would the inner formation of the ring differing be called? I know outer perimeters have names like ring, boat, chair, twist boat, etc.
Exogenous Monoamines? =
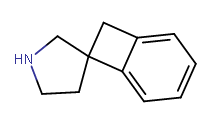
 Does cyclohexyl really qualify as a bioisostere of phenyl?..Yeah I think it does depend, and all the variables must be checked. The phenyl you substituted in diphenidine serves too much of a different role than the phenyl in amphetamine, I believe. Probably for that reason yes ^ sounds agreeable adder.
Does cyclohexyl really qualify as a bioisostere of phenyl?..Yeah I think it does depend, and all the variables must be checked. The phenyl you substituted in diphenidine serves too much of a different role than the phenyl in amphetamine, I believe. Probably for that reason yes ^ sounds agreeable adder.
It seems to me that it would be so offensive to the NMDA pharmacophore that there is either a loss of potency or overall function to be expected from it....
....I'm not sure if I'm right about this, mind you, but I think you can't always substitute with a cyclohexane or pip, I think you can't if the aromatic function is already a type of partial bioisostere itself.
IMHO The whole concept of bioisoterism should be taken with a pinch of salt. It all depends on what one is trying to achieve: eg
beat the law while retaining all biological properties of the drug (this is the easiest: just add a methyl!)
beat the competition as with big Pharma me-too drugs
increase affinity and/or target selectivity, modify PK parameters...etc etc
For Phe-->Cyclhx replacement, binding pockets of simple Phenyl of the drug on its target protein can accomodate a cyclohexyl in general. Why? because main types of interactions involving simple Phenyl (logP = 1.9) are hydrophobic in nature so yeah the cyclohexyl (log p =2.6 ) can satisfy that. even better since it is bit more interactions. Sometimes pi-stacking and/or charge transfer may also contribute like when the pocket is made-up of aromatic side chain residues of the protein. But in general the gain/loss in affinity is not a lot (+/- 2xKi may be 10x at most of the parent). So depending on what one is trying to do, one one can live with that. With a cyclohexyl, steric consideration also may play a role..(this is probably most crucial factor ie if the pocket is a bit tight to fit since the cyclohexyl is not as flat as Ph.
With AMPH and Propylhexedrine, I dont know the detail (like SAR..etc). It was just an example that came from top of my head). With NMDA antagonist
Functional NMDA antagonists like the ACA's are modelled on NMDA and electronically mimic the carbonic acid functions
That prove the point I was trying to make: it really depends. Nobody would have thought of a 3-Methoxyphenyl as a replacement for the propionic side chain of NMDA!! Whoever first discover that, I bet you, it was pure serendipity. just like 80% of the drug discovery business. Random screening, not that the guy woke up one day and said I am gonna put a 3-methoxyphenyl as a bioisteric replacement of NMDA COOH and see what happen.
That means the site where the NMDA COOH binds is rather either promiscious or it doesnt contribute much in term of the affinity of NMDA to its receptor. May be it does in terms of recognition of the substrate by the receptor ie thermodynamic vs kinetic control of the binding.. but we're getting too far in speculations re: phenyl replacement of diphenidine.
I guess bottom line is If that site can accomodate a Ph it can potentially also accomodate a cyclohexyl...but each SAR is rather unique so who knows?
It work with a 2-pyridyl :
What do they mean by "low-trapping NMDAr anatagonist? slow binding: k(on)<<k(off) ? I don't get it!!Lanicemine differs from ketamine in that it is a low-trapping NMDA receptor antagonist, showing similar rapid-acting antidepressant effects to ketamine in clinical trials but with little or no psychotomimetic side effectsI doubt that it is really coincidental about the NMDA backbone but okay. Unfortunately the PCP site is an orphan site, and yes let it be clear that we are speculating.. No NMDA is not the target ligand and no dissociatives are not exact bioisosteres of it; personally I'd expect something along the lines of the middle part being rather NMDA-like but the terminal sites having a certain electronic / aromatic interaction to change the conformation of the channel for NMDA trapping. But its just a fantasy of mine. How is it not suspicious though, that nearly all dissociatives conform to that NMDA similarity but with the terminal moieties having different properties than the carboxyls. The question would be: different how, and what is the tolerance there sterically and electronically. (It would be a huge help if someone could illustrate this with some SAR.
A side-question about this whole ordeal: would ketamine with a phenyl instead of the cyclohexane work just as well? If not, why not? The why not would be relevant to my point here.
The main thing about the pip was that it seems like it has properties unlike other moieties found right about there in other dissociatives. For all we know it could work, and yes clearly pip is a bioisostere of phenyl in a number of ways. Still, the novelty of it made me curious which is why I skeptically tried to ask about it being consistent with other NMDA antagonists. Plenty of modifications could probably be made that defy the current model we have of what NMDA antagonists look like. What I want to know is: are the reasons for expecting it to behave similarly good reasons?Last edited:
belligerent drunk
Bluelight Crew
- Joined
- Sep 16, 2015
- Messages
- 3,482
If you want to replace the cyclohexyl in ketamine with a phenyl ring, then it can't have the keto moiety. PCP doesn't have it, but even if you replace the cyclohexyl with a phenyl in PCP, then the nitrogen would be much less basic (aniline derivative), and possibly not protonated enough at physiological pH. Not sure how this would affect binding and activation, or lack thereof. If there was none of these problems, then a phenyl ring could substitute for a cyclohexyl - electronically they're not that much different, phenyl is a little bit more polar and polarizeable; however, sterically a phenyl is planar, whereas cyclohexyl tends to be bent (boat/chair conformations).
pharmakos
Bluelighter
is that just an artifact from bad synthesis?- Status
- Not open for further replies.