-
Neuroscience & Pharmacology Discussion Welcome Guest
Posting Rules Bluelight Rules Recent Journal Articles Chemistry Mega-Thread FREE Chemistry Databases! Self-Education Guide -
N&PD Moderators: Skorpio
You are using an out of date browser. It may not display this or other websites correctly.
You should upgrade or use an alternative browser.I Like to Draw Pictures of Random Molecules
- Thread starter nuke
- Start date
- Status
- Not open for further replies.



Your sentence doesn't make any sence.I have a bunch nitrimetazepams/temazepams which could fix is main weakness. It's too weak to survive as a rc.
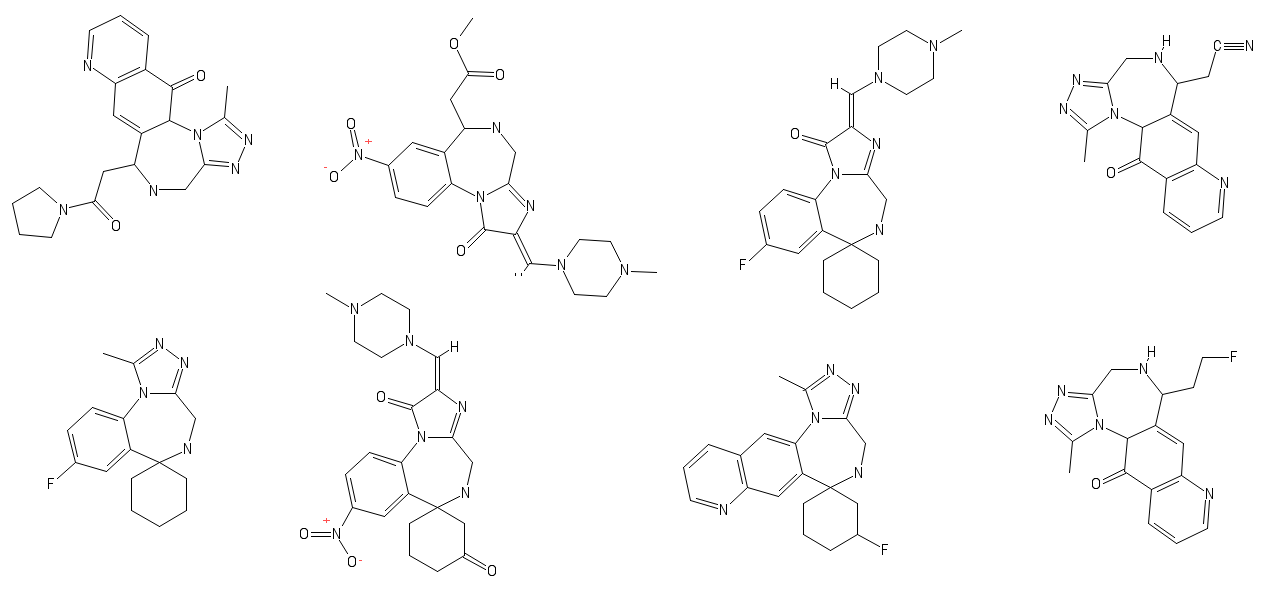
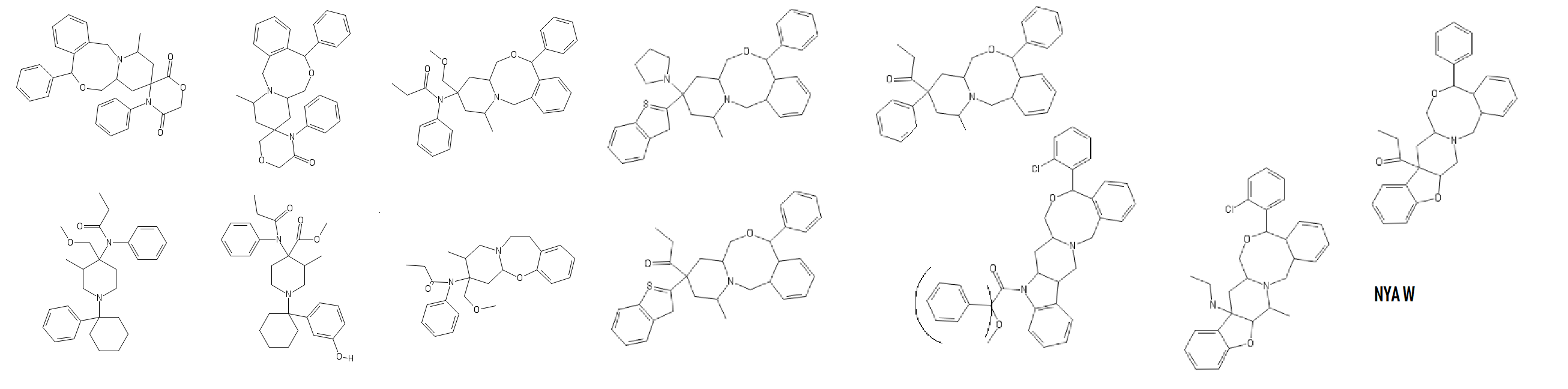
Would this be active:
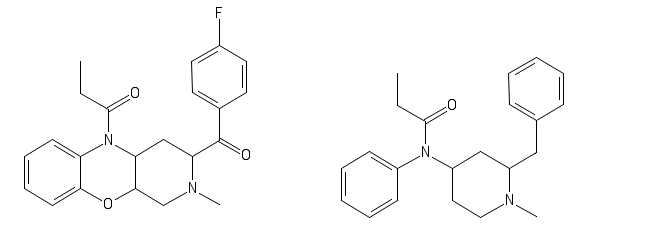
Also new, turns out Fentanyl just had a baby with Nefopam; progeniture seems wonderful:
 Last edited by a moderator:
Last edited by a moderator:
sekio
Bluelight Crew
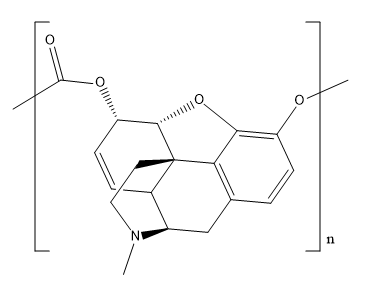
Came across the structure of viscidone, a prenylated acetophenone found in bee propolis, and, well, I had fun with Chemdraw.
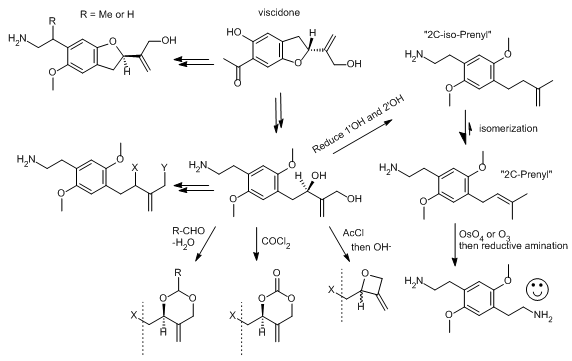
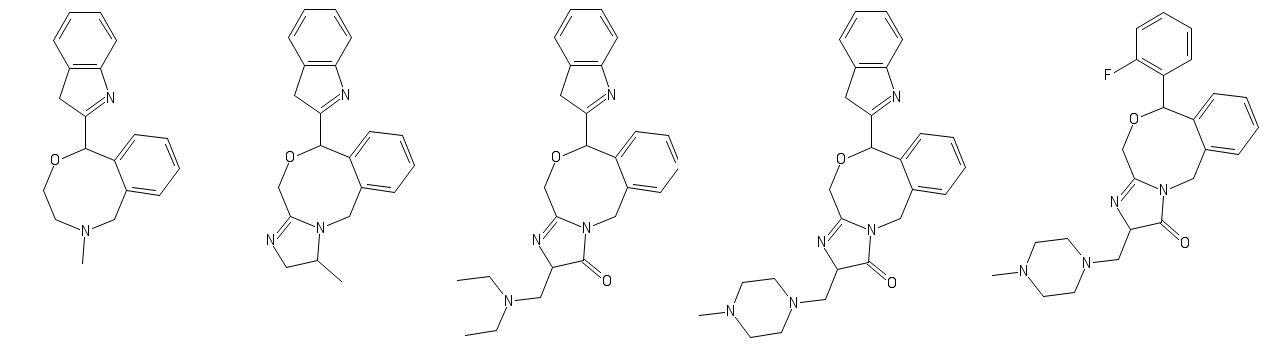
The 2C-methyl-(methyleneoxetane) is a cool idea, even if not active. And 2C-Prenyl or 2C-isoPrenyl should have been made before, even if they are low activity. Compare methallylescaline to 2C-isoPrenyl:
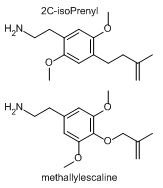 Last edited:Sekio / Dresden / Pomzazed (or anyone else reading this) do you know of a program that runs on windows that is used to map the activity of chemicals in the brain. I heard some of these exist; I'd like to get a better insight as to how active and good or inactive/poisonous the compounds I've drawn these last few days are.
Last edited:Sekio / Dresden / Pomzazed (or anyone else reading this) do you know of a program that runs on windows that is used to map the activity of chemicals in the brain. I heard some of these exist; I'd like to get a better insight as to how active and good or inactive/poisonous the compounds I've drawn these last few days are.
sekio
Bluelight Crew
There's no such thing as a "magic SAR prediction black box' whede you feed it structures and it comes back with affinity/efficacy data, unfortunately... most of the predictioms in this thread are the work of human minds drawing conclusions from already published data on drug SAR/effects.
You can find molecular dynamics or protien-ligand docking calculation software like CHARMM or GROMACS. However those programs are meant for situations where you already have a high accuracy model of the receptor in question, preferably from X-ray diffraction studies of the receptor in crystalline form. However because many receptors are typically anchored to the cell wall it can be an uphill journey to find conditions that allow a technician to both isolate the receptor in question as well as support the growth of flawless homogenous crystals of your receptor protien (plus bound ligand) that are big enough to handle and image with a cyclotron. A lot of effort is being made to derive means for recovering accurate protien structures in the solution phase just because crystallization of protiens is a black art at the best of times and seemingly impossible in the harder cases.
If you don't have the luck to have access to a good model of your target receptor, you can find a homologous receptor with a solved tertiary structure that shares a significant amount of its amino acid sequence witn your target and modify the structure accordingly, working on the assumption that the structural differences don't wildly change the shape of the receptor. I recall seeing quite a few models of different GPCRs that were based on the structure of bovine rhodopsin, for instance, because it was one of the few GPCRs people could get reasonably pure and coax into a cryatalline form.
As a last resort there is also de novo protein folding, like what Folding@Home does. However, given that protien folding is NP-complete (easy to verify a correct solution, but incredibly difficult to come up with a solution), this method takes an incredible amount of time and computational power to produce results even for protiens of relatively short length. The amount of possible configurations for each amino acid grows exponentially with every additional one so a GPCR with a few hundred to a thousand amino acids would take longer than the age of the universe to exhaust every potential conformation.
So anyway, unless you have a set of accurate models for all the receptors common to psychoactive drug effects, you're mlre likely to only be able to come up with the predicted affinity at a few receptors. Even then you'd need to mess with some pretty fancy software meant for academics and invsst some serious CPU time.Damn. Thinking of it they indeed do not know the exact sequence of most of the receptors mapping our brain, other than DA NE SER MOR Etc.. At least these are the ones I'd like to study qwa affinity vs some of the chems I drew, like the MT antagonists or some of the opioids, even if it isn't telling me the exact affinity etc, at least get an idea of how it binds to the AA sequence.
Thanks for your insight, I'll take a look at it when I'll get home from vacation.Limpet_Chicken
Bluelighter
Hell, even MOR, there are MOR1/2/3 subtypes, and a LOT of variety by ways of splice variants, potentially heterodimers or other heterooligomeric receptors between MOR and other receptors. As well as biased ligands, such as those activating strong G-protein signalling whilst producing little beta-arrestin II recruitment, the latter having a lot to blame for causing tolerance troubles.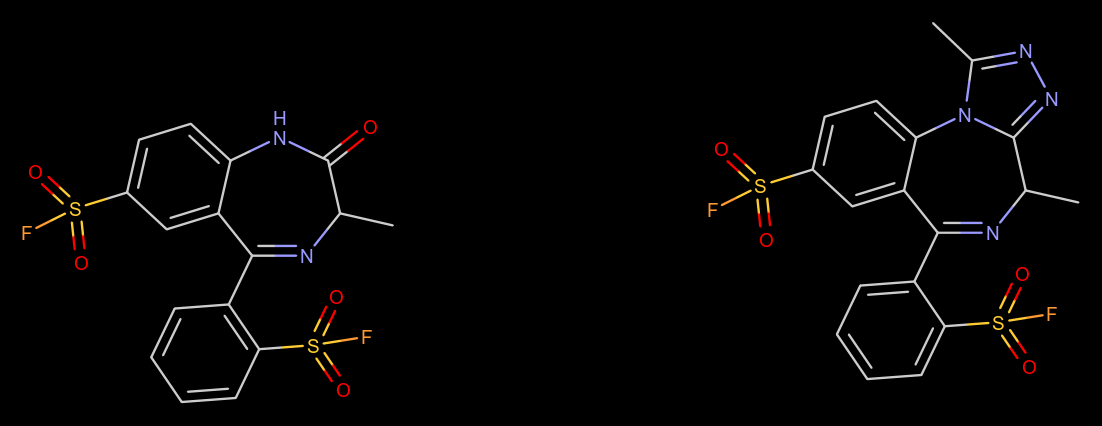
some unstandard benzo subs, do you think they would be active/commercially viable to make?
Sulfonyl fluorides tend to form covalent bonds with certain amino acid residues.
This makes them potentially extremely useful as irreversible enzyme inhibitors or dye probes in biochemistry (the field of applying such reactions which are highly energetically favorable yet selective, and which will not violently decompose in an aqueous environment, is called "click chemistry"), although it would probably make more sense to use a single rather than two sulfonyl fluoride groups; in that case, it might actually be a valuable tool for studying the structure of the benzodiazepine binding site on the various GABA receptor subunits.
https://www.sigmaaldrich.com/techni...technology-spotlights/sulfonyl-fluorides.html
But as a pharmaceutical that people would actually ingest to produce a psychotropic effect? Not really.Like this:
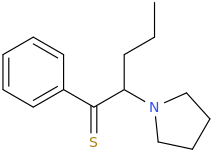
Aren't thioketones highly unstable?
Thiobenzophenone is one of the few stable ones, although there the thiocarbonyl is benzylic to not one, but two benzene rings.
Still, even that one tends to readily get photooxidized (into sulfur and benzophenone).
Not that anyone would ever willingly ingest a thioketone. I am sure Limpet Chicken can come up with a graphic description of what this stuff must smell like, and is consequently going to make the user smell like ("It's like Andrew Wakefield pooping on Margaret Thatcher's rotting corpse while Jacob Rees-Mogg sits in the background, masturbating and farting.")") During my M.Sc. I have synthesized thioketone deriv. of JWH-018
During my M.Sc. I have synthesized thioketone deriv. of JWH-018
it is morr stable than i predicted, it survives air and room temp for many days,
it forms a black solid and deep red solution in DCM too.
That's pretty cool.
Still, "thio-JWH-018" has the advantage of having that thiocarbonyl next to not just one, but two aromatic systems (naphthalene and indole), similar to thiobenzophenone's two phenyl rings.
I wonder how stable something with just a single aromatic system on the alpha carbon could be.
Benzylthio like in BTCP, with the ketone on the carbon above the S group
There is no benzylthio in BTCP. Did you mean benzothiophene?
Also... where is "above"?
Skeletal formulas can be rotated any which way; you need to post the one you mean.
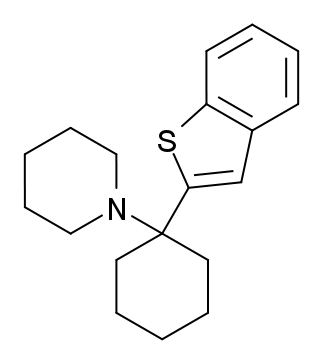
Is it this one?
Because the carbon "above" the sulfur atom here cannot accept any more bonds.Last edited by a moderator:Enough joking, I was indeed wrong about the spelling. What I meant was benzothiophene-PVP, like in methiopropamine with a benzene ring added to the 3-4 position.
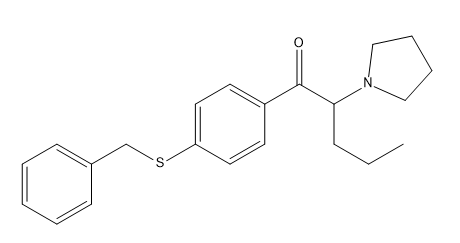
You mean 1(2-benzothiophenyl)-2-pyrrolidino-pentan-1-one ?
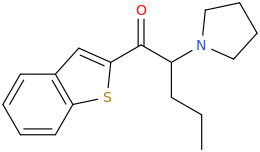
Edit: You said 3-4 position, so maybe this one?:
1(3-isobenzothiophenyl)-2-pyrrolidino-pentan-1-one
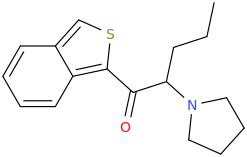 Last edited:
Last edited: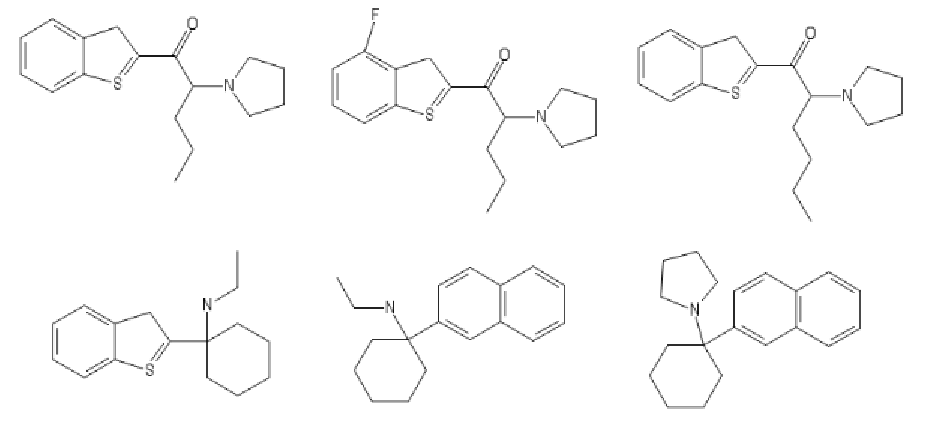
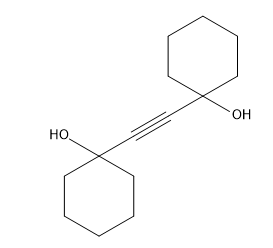
These are the ones I had in mind. The double carbon bond should be at the sulfur, makes a more stable compound.
I know it's merged, please Don't edit; shouldn't have been done on the "Hodor" comment, lost its humor.Last edited:- Status
- Not open for further replies.