Wow, really? I've actually tried taking 900mg of clobazam at once and the side-effects were no greater than those delivered by 40mg/day. Sure, the duration of action was MUCH longer because even after days, their was still more than the ceiling dose in my body.
I have posted hotlinks to the papers that demonstrated the ceiling in a pretty robust manner. Maybe it's some specific condition (not identified in the 70s) that benefits from 80mg/day but that makes it a great drug - the side effects have a much lower ceiling than the activity... which is something I've only seen in PARTIAL agonists. So that's a useful insight for me, thank you
@emkee_reinvented
While studying the design of the alcohol mimic, we had several novel compounds called 'the supercats' (because our novel 1,4-benzodiazopines were named after our cats (Sniper = pyrazolam, Lettie = Pynazolam & Sally = pyeyzolam) so any intercepted E-mails wouldn't identify what was being discussed. I thought it silly, but Nutt insisted).
Image 1 hosted in ImgBB

ibb.co
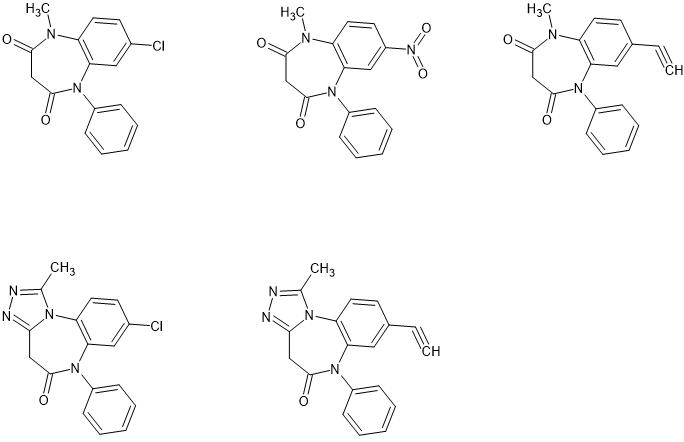
So top-left is our precursor (clobazam). There are name-reactions to concert the 7-Cl to a nitro (Friedel-Craft) or an ethynyl (Suzuki coupling) in 1-pot techniques, we didn't need a good yield or worry about side-reactions because preparative chromatography was used in all cases and if it cost $2000 to buy 500 grams of clobazam, it saved >$2000 in designing and optimizing a specific route.
Now nitrazam was certainly more sedating but of more interest was that it wasn't a potent seorotonin-releasing agent as pynazolam proved to be. Etylazam WAS alcohol-like. Importantly, it was able to mimic slight intoxication. The big problem wasn't that pyeyzolam wasn't good at simulating alcohol intoxication.. the problem that it was TOO good. If a person swallowed 20mg (as a 1mg/mL solution) their was barely an effect... but if that same person swallowed 30mg a week later (to ensure first dose has been entirely excreted) then they would be VERY 'drunk'.
But then we did get interested in finding out if it were possible to produce a more active version of etylazam. So we produced the triazolo derivative of clobazam and it proved to be around x4 more potent - not the x10 exhibited in the 1,4-benzodiazepines. But subjectively the product was very similar to clobazam, a reduced duration of action due to it being excreted unchanged.
Now we did then swap the halide for an ethynyl to make pyeyzam. I never got to see the instrumental data on that one. I was just sent a 1.5L plastic bottle with the legend 'Super Sally - 1mg/ml + 3mL/mL' which proved to be a superb base for cocktails. A phone-call confirmed that it should simply be treated like any clear alcoholic spirit.
Then we ran into the financial hurdles. We HAD a product that accurately simulated all of the positive effects (relaxation, increased happiness, reduction in anxiety, disinhibition and so on) of alcohol without any of the negative effects (mood lability, increased aggression, retrograde amnesia, anteriorgrade amnesia, nausea, ataxia, increased urination leading to dehydration, loss of judgement & so on). As far as alcohol's effects on the brain, (almost) all of the positive effects are mediated by the a5 subunit and (almost) all of the negative effects by the a1 subunit.
That's because pyeyzolam is a5b1y2 selective while pyeyzam is a5b2y2 and a5b3y2 selective - the mixture of two compounds produced a drug that was overall LESS selective but importantly, avoided a1 activity which seems to be a key element in physical dependence of alcohol developing as well as some of the negative effects alcohol causes.
But then we reached the position where, even when we provided our boss with the estimates for the cost of developing the mixture, it proved to be a difficult problem. Because the only legal way to introduce a mind-altering drug we knew of was to have it licenced as a medicine, any mixture would double the risks, double the costs and potentially double the time. Our aim was simply to get it accepted as a medicine to treat dependent alcohol users. We even found that Tipplersbane mushrooms contain a natural compound that is an aldehyde dehydrogenase inhibitor which we intended to add as in effect, it's action is like that of Antabuse (disulfiram). It felt like we had it and it would all become easier after three years of hard work.
After a few months were informed that financially, we HAD to find a single compounds that would substitute for the 2 candidates we had already found. That was a shock. The earlier stuff was all based on the research of others (who are cited in the patent) but this was something for which their was no known scaffold that would allow for a selectivity but had no beta selectivity.
We spent about 6 months researching all the papers that used the kavalactones and various other natural intoxicants first to find the subset whose effects were mediated by GABA activity and then we went through every paper and patent that dealt with anxiolytics, hypnotics and even anti-epileptics. We kept on expanding and expanding the search... it started to look like their was no evidence that such selectivity and activity was possible with a single candidate.
")