Limpet_Chicken
Bluelighter
- Joined
- Oct 13, 2005
- Messages
- 6,323
Can anyone theorize on the activity of the following two compounds:
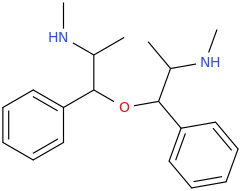
Pseudoephedrine-PFED dimer, linked by an ether bridge after dehydration-condensation of the two beta-OH groups.
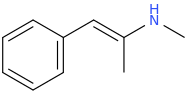
Pseudoephedrine, having either lost the OH or not, but elimination to a beta-gamma alkene.
At least one of these SEEMS to be active as a psychostimulant, expected methamphetamine, but the synthesis used, reflux with sodium (meta)bisulfite/bisulfite in aqueous conditions for several hours (longer than the etherification takes going from a research journal article, will post when I can) seems instead to have produced a mixture of the alkene and the symmetrical diether.
Dose-unknown, a large aliquot of the post-rxn workups aqueous phase was taken, the naphtha removed via sep funnel, evaporated a little off the top to remove as much as possible of the nonpolar phase, at this point the results of the rxn were in the aq. phase as the hydrochloride salts. PH neutralized to made slightly basic with sodium bicarbonate, then plugged.
This was primarily a test to discern weather or not pseudo was present in any great degree for it makes me quite ill, and very badly overloaded, so I decided to use that as a marker of biological activity.
Instead of triggering a massive overload (I.e of the autie kind)
But instead of making me ill and feel like shite on a burger bun I nevertheless ended up flying for quite a long time, had to wait overnight to pick up a repeat prescription so did an allnighter, but the time positively shot by. Redosed a couple of times with a lesser amount, although unknown.
The active fraction whatever it may be, appears not to be quite so forcefully dopaminergic as methamphetamine, but nor, is it so strongly (nor)adrenergic, this observation however must be taken in context of it coming from bioassay data. However I CAN'T tolerate pseudoephedrine or ephedrine at all systemically, I can do so as a nasal decongestant but only extracted from pills then redissolved in distilled water and taken a drop or two up each nostril in dilute solution, this because I cannot tolerate a full dose via tablet form orally (30mg)
Whatever the dose was, it was fairly large, but definitely active psychostimulant, and LONG acting, longer than meth. I can still feel it a little. After initially 8 hours or thereabouts, I had to turn things down a notch by means of 400mg chlormethiazole, a large dose of oxy plus a little short of 0.5g morphia via the IV route, some clonidine and tizanidine. This combination proved sufficient to induce sleep, just about, although needed a further dose of chlormethiazole a bit later on. I STILL feel wide awake and motivated, and the drug was taken on late sunday night, having been slaving at the rxn for quite some time, eventually got done more or less late at night.
Results were so pronouncedly active that whilst I was shot after all the time spent babysitting the rxn, I was tired, hot, cranky and thirsty. So I cracked a cold beer, but proved unable, after dosing, to take more than two swallows, appetite just up and vacated.
Pseudoephedrine-PFED dimer, linked by an ether bridge after dehydration-condensation of the two beta-OH groups.
Pseudoephedrine, having either lost the OH or not, but elimination to a beta-gamma alkene.
At least one of these SEEMS to be active as a psychostimulant, expected methamphetamine, but the synthesis used, reflux with sodium (meta)bisulfite/bisulfite in aqueous conditions for several hours (longer than the etherification takes going from a research journal article, will post when I can) seems instead to have produced a mixture of the alkene and the symmetrical diether.
Dose-unknown, a large aliquot of the post-rxn workups aqueous phase was taken, the naphtha removed via sep funnel, evaporated a little off the top to remove as much as possible of the nonpolar phase, at this point the results of the rxn were in the aq. phase as the hydrochloride salts. PH neutralized to made slightly basic with sodium bicarbonate, then plugged.
This was primarily a test to discern weather or not pseudo was present in any great degree for it makes me quite ill, and very badly overloaded, so I decided to use that as a marker of biological activity.
Instead of triggering a massive overload (I.e of the autie kind)
But instead of making me ill and feel like shite on a burger bun I nevertheless ended up flying for quite a long time, had to wait overnight to pick up a repeat prescription so did an allnighter, but the time positively shot by. Redosed a couple of times with a lesser amount, although unknown.
The active fraction whatever it may be, appears not to be quite so forcefully dopaminergic as methamphetamine, but nor, is it so strongly (nor)adrenergic, this observation however must be taken in context of it coming from bioassay data. However I CAN'T tolerate pseudoephedrine or ephedrine at all systemically, I can do so as a nasal decongestant but only extracted from pills then redissolved in distilled water and taken a drop or two up each nostril in dilute solution, this because I cannot tolerate a full dose via tablet form orally (30mg)
Whatever the dose was, it was fairly large, but definitely active psychostimulant, and LONG acting, longer than meth. I can still feel it a little. After initially 8 hours or thereabouts, I had to turn things down a notch by means of 400mg chlormethiazole, a large dose of oxy plus a little short of 0.5g morphia via the IV route, some clonidine and tizanidine. This combination proved sufficient to induce sleep, just about, although needed a further dose of chlormethiazole a bit later on. I STILL feel wide awake and motivated, and the drug was taken on late sunday night, having been slaving at the rxn for quite some time, eventually got done more or less late at night.
Results were so pronouncedly active that whilst I was shot after all the time spent babysitting the rxn, I was tired, hot, cranky and thirsty. So I cracked a cold beer, but proved unable, after dosing, to take more than two swallows, appetite just up and vacated.
Last edited: