Image 1 hosted in ImgBB

ibb.co
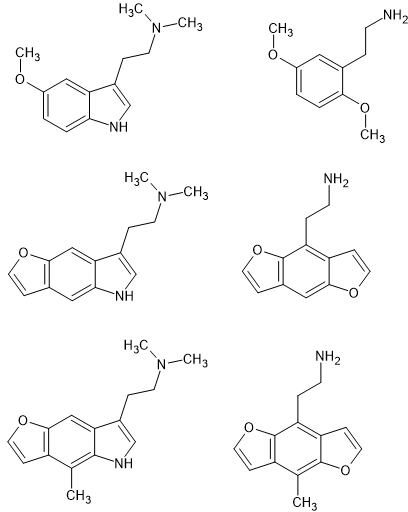
I thought an image would be much more telling than a bunch of SMILES, IUPAC or IChi texts.
As you can see, the N of the pyrrole portion of an indolve overlays the 5-MeO of 2,5-dimethoxy-4<something> PEAS.
It's also worth noting that the double-bond is important. 5/6APB is more potent than the 5/6MAPB counterparts because the O/N is planer to the benzene ring.
So that explains why 5-MeO indoles are so much more potent (and other effects make them toxic - best avoided) AFAI know nobody had made the 6-APB homologue of indoles - but that might be synthetic complexity.
So IS it a surprise that 6-methyl indoles are avtive since 5-methoxy-6-methyl amphetamine was detected in Italy in the 90s in dose-units of 141mg (mean.
So, going further, no surprise that 2-(8-methylbenzo[1,2-b:4,5-b']difuran-4-yl)ethan-1-amine (2-(8-Methylfuro[2,3-f][1]benzofuran-4-yl)ethanamine) is known, WO2022006186 - PHENALKYLAMINES AND METHODS OF TREATING MOOD DISORDERS
In short, the insoles and the PEAs are very closely related in their binding and action.
I admit I forget the name but their are NBome derivatives of both PEAS and tryptamines which is another god clue to the fact that they bind in the same manner.
Now what interests me is IF it's possible to produce an N-bom derivative of aminorex and it is possible. Well, some idiot made 2,5-diemthoxy-4-bromo aminorex.... which amazingly wasn't active. No surprise there because it's a 5HT2a ligand.
BUT what is more interesting, it if it's possible to use the N-bomb route as a methodology to provide boarderline 5HT2a ligands (active but lacks sufficient affinity to be active at practical doses. So if anyone can suggest compounds with mild activity but require a LOT, maybe it's worth considering.
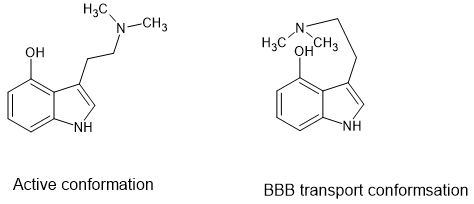
I will finish with the common question 'why is 4-hydroxy DMT active but 5-hydroxy DMT is not. The reason is simple, the H of the hydroxy will form a hydrogen-bond with the amine function so that it can assume a form that makes it active. I have indeed considered if their are better alternatives but nature does a great job.
Image 1 hosted in ImgBB
ibb.co
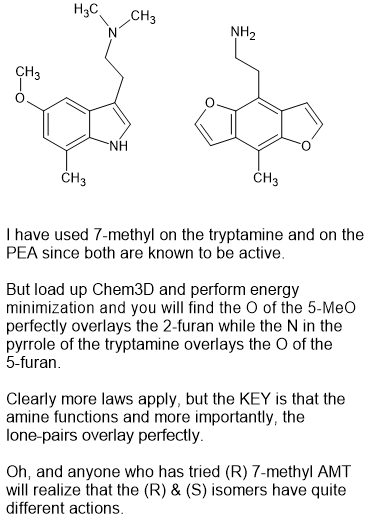
Sorry for my crappy drawing but you get the idea.
It's SUCH a pain that production if 6-methyl indoles is such a pain. Because trust me. It's a very special class of compound. It has all of the warmth of things like psilocin but it releases dopamine as well as serotonin so the subjective effects really ate excellent, You see, the 6-methyl acts like the p-Me of PEAs so it's a little like p-TAB but because of the serotonin, you do not need LOTS of dopamine.
Over 2 years I researched over 100 compounds and 70 were dumped within 2 days because of the cost... or rather cost per dose. U-47700 was more costly because (1R,2R)-N1,N1,N2-trimethylcyclohexane-1,2-diamine (CAS: 67198-26-9) but at £60/g and the other 2 precursors being off-the-shelf, it all worked out.
BUT anyone making it will know that (1R,2R)-N1,N1-dimethylcyclohexane-1,2-diamine is the intermediate that is N-methylated BUT one only has to look at related compounds with groups in place of the N-methyl. I mean, Take a look at the most active form of allylprodine (3R,4S) (z23 M) and then try replacing the N-methyl with an N-allyl. Their are 4 isomers of allylprodine, only 2 of U-47700 so presuming one is x.7M, the other is x23 and you wind up with 12.5M.
If you are smart enough to resolve, it's going to be x23.
")