

Harm Reduction Question: Does anybody heard about this compound?
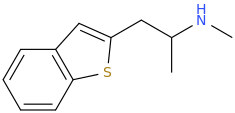
it is a cousin of MDMA
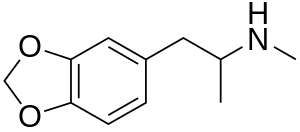
with the 3,4methylenedioxyphenyl replaced by a benzothiophene. Reportedly it is empathogen with "exact pharmacology" as MDMA but 2x as potent (from vendors so to be taken with a pinch of salt!!!) and not neurotoxic unlike MDMA. I know that benzothiophenes are pretty safe as far as oxidative metabolism-induced neurotoxicicty is concerned. Actually they are neuroprotectants, antioxidants like glutathione (ref later): sulfur (sp3 as in glutathione) inhibits oxidative metabolism that generates reactive oxygen species that lead ultimately to neurons death. IIRC, the consensus of MDMA neurotoxicty is that it gets metabolized to reactive toxic 1,2 quinones which generates reactive oxygen species leading to cell death. Benzothiophene won't get metabolized similarly so it would make sense this compound could be non neurotoxic I mean at least as far oxidative metabolism -induced neuronal cells damage is concerned.
nb: would be interesting, for the sake of harm reduction, but I have yet to see any data on that molecule.. lemme me know if anybody came across this compound/data/experience..etc .. thx
it is a cousin of MDMA
with the 3,4methylenedioxyphenyl replaced by a benzothiophene. Reportedly it is empathogen with "exact pharmacology" as MDMA but 2x as potent (from vendors so to be taken with a pinch of salt!!!) and not neurotoxic unlike MDMA. I know that benzothiophenes are pretty safe as far as oxidative metabolism-induced neurotoxicicty is concerned. Actually they are neuroprotectants, antioxidants like glutathione (ref later): sulfur (sp3 as in glutathione) inhibits oxidative metabolism that generates reactive oxygen species that lead ultimately to neurons death. IIRC, the consensus of MDMA neurotoxicty is that it gets metabolized to reactive toxic 1,2 quinones which generates reactive oxygen species leading to cell death. Benzothiophene won't get metabolized similarly so it would make sense this compound could be non neurotoxic I mean at least as far oxidative metabolism -induced neuronal cells damage is concerned.
nb: would be interesting, for the sake of harm reduction, but I have yet to see any data on that molecule.. lemme me know if anybody came across this compound/data/experience..etc .. thx
