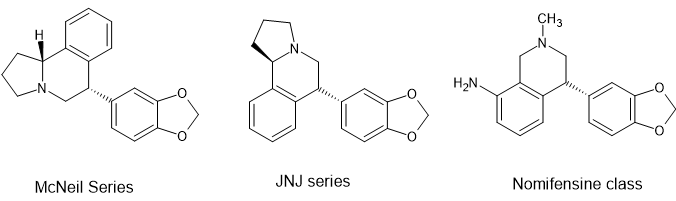
Dear Bluelight,
I'm a PhD student, soon doctor if I don't die in the process before the end haha. I would like to know if some people here are working on in silico drug design, and more generally molecular modelling and also machine learning approaches?
I love my job. Molecular modelling is so cool. It's so beautiful to look at the atomic level how your receptor behave. I would like to share this a bit with you bluelighters. Just to start and see how to put images, a little GIF of a zoom into the LSD bound 5HT2B receptor crystallographic structure, at the binding site level. The LSD is in magenta, and amino acids of 5HT2B distant from ~3 A or less are in green. You can see the interaction with dashed lines. You have some hydrophobic ones, some ionic bound (hallmark of aminergic classA GPCRs), some T stacking...
I'm a PhD student, soon doctor if I don't die in the process before the end haha. I would like to know if some people here are working on in silico drug design, and more generally molecular modelling and also machine learning approaches?
I love my job. Molecular modelling is so cool. It's so beautiful to look at the atomic level how your receptor behave. I would like to share this a bit with you bluelighters. Just to start and see how to put images, a little GIF of a zoom into the LSD bound 5HT2B receptor crystallographic structure, at the binding site level. The LSD is in magenta, and amino acids of 5HT2B distant from ~3 A or less are in green. You can see the interaction with dashed lines. You have some hydrophobic ones, some ionic bound (hallmark of aminergic classA GPCRs), some T stacking...

Last edited:
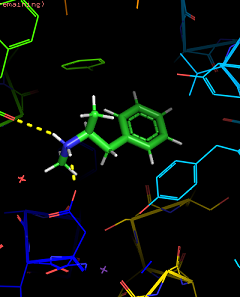
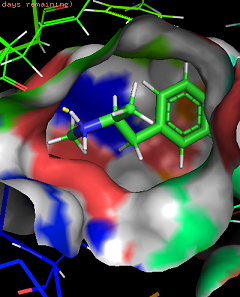
")