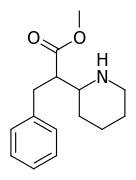
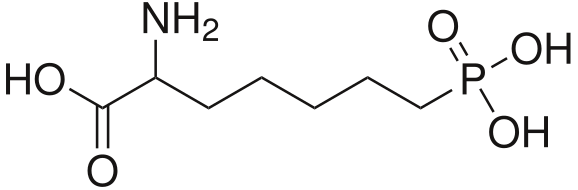
It would, but it doesn't seem likely: in ACA's the optimal length is ethyl, longer than that (besides constraining in heteroring) and they simply get less potent. NMDA is an excitotoxin, if any of the SAR makes any sense a lengthy alkyl chain won't cause the aspartic acid derivative to bind without activation (i.e. antagonistic), but just have less affinity so a little less toxicity.
I realize there may be a different ruleset for antagonists, but at least for ACA's the pharmacophore and similarity to NMDA should be a match?
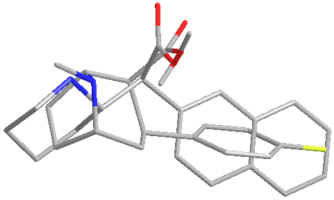
I saw an image of a overlay of an NMDA molecule overlaid a methoxetamine molecule and had this idea. Cannot find the original image at this point I lack good tools to explore this further but I am very curious.
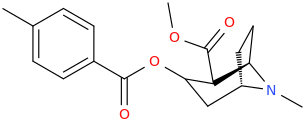
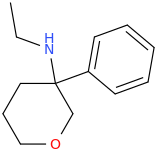
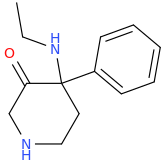
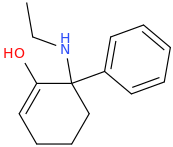
Not a bad idea, I've fantasized about different ways to mimick NMDA than the typical groups in arylcyclohexylamines, but not what you posted.
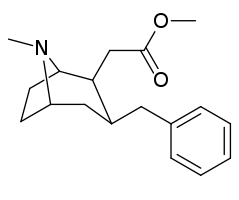
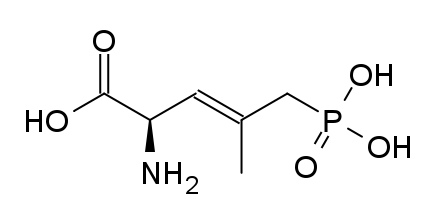
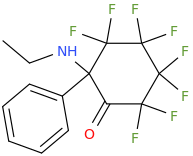
However counting from one carboxylic acid to another, you seem to have one added spacer when comparing to NMDA, so that =O oxo group you have on 3 (a la MXE) would have to be on 2-position now. In MXE and other arylcyclohexylamines part of the phenyl ring (from the 2-position to the 3-position) plays a role similar to that of the oxo.
Now that you made the phenyl ring into a cyclohexyl ring, that is not true anymore, so the electron-rich moiety will now have to stick out, but closer by than the 3 position.
Am I explaining my idea well or should I draw it? (Not long ago in this thread molecules were posted that intentionally had different spacing - more specifically of the amine position compared to the carboxylic acid like functions, and who knows if some of these would work)
Another thing is that I would not be surprised if the arylcyclohexylamines work well in part because of an aromatic site in the channel that pi-stacks with the phenyl ring, good for sticky binding and coordination. Off the top of my head, MK-801 ketamine DXM diphenidine they are all aromatic.
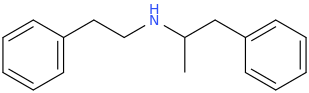

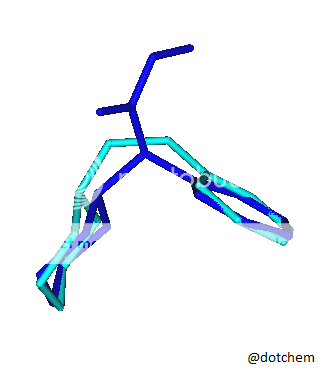
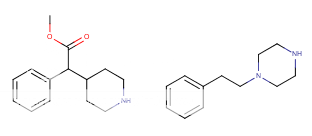
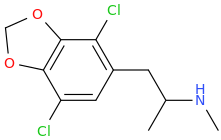
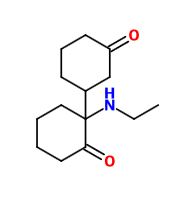

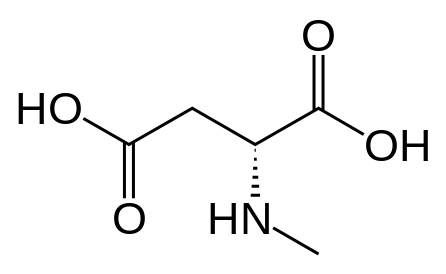
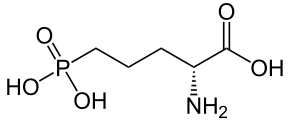
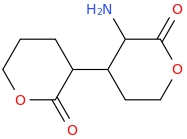
![3-amino-3-[%282-oxooxan-3-yl%29methyl]oxan-2-one.png](/community/proxy.php?image=http%3A%2F%2Fopsin.ch.cam.ac.uk%2Fopsin%2F3-amino-3-%5B%25282-oxooxan-3-yl%2529methyl%5Doxan-2-one.png&hash=0bcd8da38218776495b36ce53ecebd57)