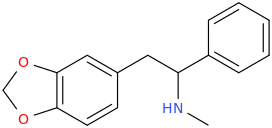
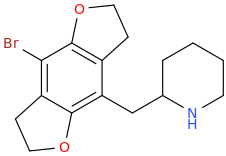
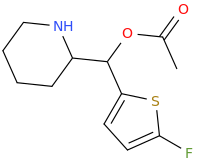
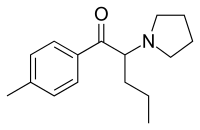
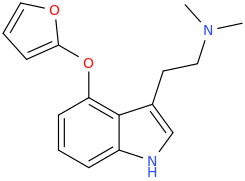
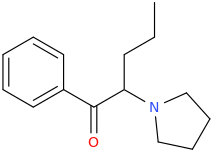
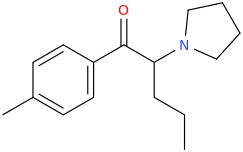
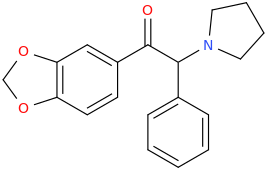
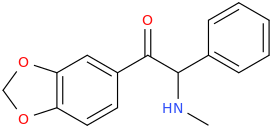
It seems counter-intuitive but actually IT IS a reuptake inhibitor unlike MDMA. It reverses inward-pre-pulse synaptic current in fashion similar to Cocaine and not AMP-like releasers such as methylone which basically dump dopamine in synaptic cleft without reversing the current. The SAR (at that alfa-position) is that the more substituents you put there beyond a methyl the more cocaine-like reuptake inhibitors you end up with. This is true for both AMP-like and methylone-like(bk-AMPs.I know on Wikipedia it says it's a reuptake inhibitor but I doubt this claim...
So you got from Methylone(---->ethylone--->butylone---.pyrovalerone from (releaser ----> reuptake inhibitor). or from AMP ----> Prolintane which is more cocaine-like DRI and less AMP-like). Here is a thorough review on this question by the upmost expert on the field Pr Rick Glennon : (a gold mine to design new and improved Cocaine-like):
“Deconstruction” of the Abused Synthetic Cathinone Methylenedioxypyrovalerone (MDPV) and an Examination of Effects at the Human Dopamine Transporter ACS Chem Neurosci. 2013 Dec 18; 4(12): 1524–1529.
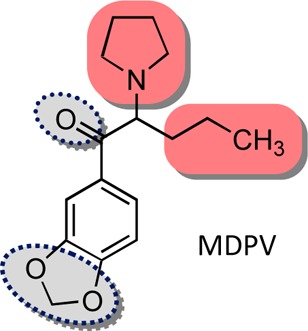
The data on UWA-101 reported by the references on the wiki entry refer to Reuptake Inhibition. Granted, we dont know whether the UWA series are also releaser as the authors didin't mention it in their abstracts but one thing is certain: their pharmacology is dramatically different from MDMA and very much closer to cocaine. Which bring back my question: Do they have anything in common(structurally, electronically...etc) with the tropanes like the RTI-83? Or may be they bind to the transporter similarly (same site and triggering same conformational change of the transporter leading to current reversal.. who knows?
Thanks @nag for pointing that out. The Ki data refer to the 4propenylphenyl tropane instead of the 4ethylbenzene dervative. It is even more intriguing now considering the rgidity put by the propenyl. if you consider the DAT and NET, (any) substitutenst that kill NET activity wil also kill DAT binding but obviously this is not the case!Those aren't RTI-83s values, those are:
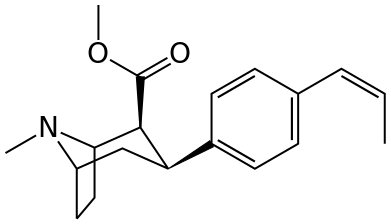
RTI-304s values. RTI-83s are:
(Ki)
55 nM @ DAT
28.4 nM @ SERT
4,030 nM @ NET
As for your question, it seems, if you look at the difference between DA & NE, that the ethyl or cis-propenyl (and in the amphetamine skeleton, the alpha methyl lengthened to a cyclopropane) somehow leaves the hydroxy group that alters NEs binding free to pry itself into the transporter that are otherwise blocked when it comes to DA or 5-HT; of course, that's an obvious non-explanation explanation, but such is QSAR.