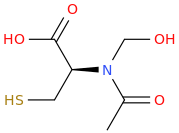
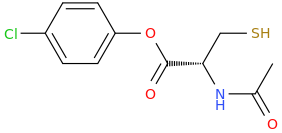
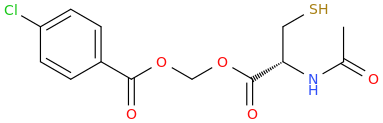
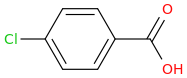
dopamimetic
Bluelighter
- Joined
- Mar 21, 2013
- Messages
- 2,070
There is very exciting new evidence for the use of N-acetylcysteine as an antioxidant with robust improvements in several neurological / psychiatric disorders like depression, anxiety, chronic stress / fatigue etc. Oh, and it was shown to protect against toxic / oxidative changes to DA neurons from methamphetamine etc. and possibly reduces tolerance development and overall after effects from psychostimulants.
Source: N-acetylcysteine in psychiatry: current therapeutic evidence and potential mechanisms of action
N-acetylcysteine (NAC) in neurological disorders: mechanisms of action and therapeutic opportunities
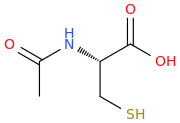
But this compound has a really poor oral bioavailability of just 9-10% which severely limits its use.
Thanks")
Source: N-acetylcysteine in psychiatry: current therapeutic evidence and potential mechanisms of action
N-acetylcysteine (NAC) in neurological disorders: mechanisms of action and therapeutic opportunities
But this compound has a really poor oral bioavailability of just 9-10% which severely limits its use.
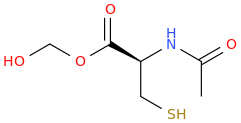
(from the second publication) How could one improve the NAC molecule to achieve a better tolerability and bioavailability, e.g. over a prodrug? And how about the BBB permeability?Low bioavailability of NAC is one of the major limitations for maximizing its effects on oxidative stress-related diseases. Giustarini et al. (2012) reported that esterification of the carboxyl group of NAC to produce N-acetylcysteine ethyl ester (NACET) would increase the lipophilicity of NAC as the mechanism of increasing its pharmacokinetics. They showed that NACET is rapidly absorbed in rats after oral administration, but reaches very low concentrations in plasma. This is due to a unique feature of NACET: it rapidly enters the cells and transforms into NAC and cysteine (Giustarini et al. 2012). After oral treatment, NACET (but not NAC) was able to increase significantly the GSH content of most tissues in the rat (including brain), and protected them from paracetamol intoxication. To overcome this limitation of NAC, an amide derivative, N-acetylcysteine amide (NACA) has been synthesized to improve its lipophilicity, membrane permeability, and antioxidant properties. Recent studies have demonstrated the blood–brain barrier permeability and therapeutic potentials of NACA in neurological disorders (Sunitha et al. 2013).
Thanks
Last edited: