mr peabody
Bluelight Crew
- Joined
- Aug 31, 2016
- Messages
- 5,714

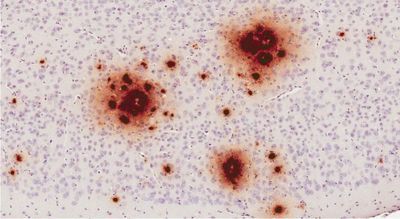
BRAIN BUILDUP Amyloid-beta (illustrated in brown) from vials of contaminated growth hormone spread in the brains of mice,
researchers found.
Alzheimer’s-related protein can be spread by tainted growth hormone
A hormone treatment contaminated with amyloid-β given to mice caused the protein’s accumulation in their brains, suggesting the same could have occurred in humans given the therapy.
Researchers injected mouse brains with a growth hormone tainted with amyloid-β, a protein implicated in Alzheimer’s disease, and saw it accumulate. The study, published yesterday (December 13) in Nature, bolsters the hypothesis that amyloid-β may be spread from one person to another under rare conditions from contamination, but does not show the protein to be contagious.
The idea that the protein could be transferred between people gained traction after a 2015 study found unusually large deposits of amyloid-β in the autopsied brains of four people who had received injections of growth hormone as children in the United Kingdom. Thousands of children received growth hormone derived from cadavers to treat stunted growth between 1958 and 1985, The Guardian reports.
The new study confirms that some growth hormone back then was contaminated with amyloid-β. The researchers then injected the contaminated hormone into the brains of mice "genetically engineered to be susceptible to amyloid pathology," according to Nature's news report. These mice grew plaques, chunks of amyloid-β protein that are found in the brain of Alzheimer’s patients. Taken together, the findings suggest that amyloid-β can “seed” the protein’s buildup in human’s brains too.
Synthetic growth hormone has since replaced that derived from cadavers. But to prevent this sort of spread in the future, The Guardian reports, surgeons should decontaminate tools used for brain surgery more thoroughly.
https://www.the-scientist.com/news-...in-can-spread-by-tainted-growth-hormone-65216
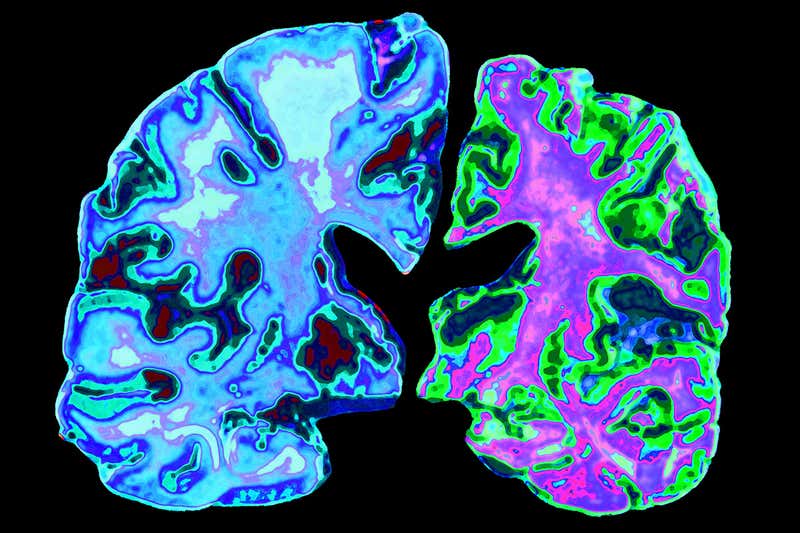
Alzheimer’s leads to the loss of brain matter (right)
Childhood hormone treatments may have spread Alzheimer’s proteins
Growth hormones given to children decades ago appear to have spread proteins linked to Alzheimer’s. The finding adds to evidence that Alzheimer’s proteins can be spread between people.
Between 1958 and 1985, approximately 30,000 children around the world received injections of human growth hormone extracted from dead bodies to treat genetic disorders and growth deficiencies.
Three years ago, while examining the brains of eight people who had received such injections and later died of the rare brain disorder Creutzfeld-Jakob disease (CJD), John Collinge at University College London and his colleagues noticed they all had beta-amyloid proteins in their brains.
Beta-amyloid is known to accumulate and form sticky plaques in the brains of people with Alzheimer’s disease. These eight people didn’t have this condition, as they all died from CJD at a young age, but Collinge says that had they lived, it’s possible that they would have gone on to develop it.
“That led us to hypothesise that the reason they got this amyloid is because those growth hormone batches that were prepared many years ago with human tissue were contaminated with this protein,” says Collinge. Before synthetic alternatives were available, human growth hormone was extracted from the pituitary glands of cadavers.
“Another suggestion was maybe it’s the growth hormone itself that stimulates the amyloid beta pathology, and not any contaminant,” he says. To investigate, Collinge and his team used samples of the human growth hormone that were given to these eight people, which had been archived by a health body in the UK.
Seeding disease
They found that all of the eight people had received growth hormone extracted from cadavers using one particular method. When Collinge and his team compared growth hormone prepared in this way with three other extraction methods, they found beta-amyloid proteins and tau proteins – which are also implicated in Alzheimer’s – only in samples made using this method.
To see if this exposure to Alzheimer’s proteins could have been enough to seed later disease, the team injected archived human growth hormone into the brains of mice that had been genetically engineered to be able to develop some human-like signs of Alzheimer’s disease.
These mice went on to accumulate beta-amyloid and tau proteins in their brains, as did mice that were injected with brain tissue from Alzheimer’s patients. Mice injected with synthetic growth hormone, however, did not show any signs of these proteins. Together, these results suggest some cadaveric formulations of growth hormone can indeed seed the accumulation of Alzheimer’s-related proteins.
“I was rather amazed that we could seed this so easily with this material that sat around as a dried powder for 30 or 40 years. It shows the persistence of these seeds to degradation,” says Collinge.
Long-lived proteins
"The persistence of beta-amyloid may explain why its build-up in the brain is so difficult to fight," says David Holtzman at Washington University in St. Louis, Missouri. “They are stable once they form, often for the lifetime of the individual. Once these things form, they continue to grow and grow, and the body can’t get rid of it easily,” he says.
While Alzheimer’s proteins seem to be transmissible, that doesn’t mean they are contagious. “You have to come in contact with brain matter,” says Sebastian Brandner, who worked on the study. “Perhaps other transmissions through other organs may be possible. There’s no evidence of that, but there’s a huge amount of research that can be done,” he says.
In a previous study, Brandner found that eight people who developed cerebral amyloid angiopathy – a build-up of beta-amyloid proteins in the brain that can lead to bleeding – at an abnormally young age, had all undergone childhood brain surgery. He found that the surgical instruments could have transmitted the proteins.
Collinge says contamination of surgical instruments is a small risk, but one we should remove through research and better sterilisation techniques.
https://www.newscientist.com/article...mers-proteins/
Herpes viruses implicated in Alzheimer’s Disease
A new study shows that the brains of Alzheimer’s disease patients have a greater viral load, while another study in mice shows infection leads to amyloid-β build up.
he brains of Alzheimer’s disease patients have an abnormal build up of amyloid-β proteins and tau tangles, which, according to many researchers, drives the ultimately fatal cognitive disease. This theory is being challenged by a newer one, which posits that microbes may trigger Alzheimer’s pathology.
Two new studies, using different approaches, further bolster this pathogen theory. Analyzing the transcriptomes of post-mortem brain samples from patients with Alzheimer’s disease, one group of researchers finds that two strains of human herpesvirus are significantly more abundant than in the brains of people of the same age without Alzheimer’s disease. Gene networks in the brains of Alzheimer’s patients with these strains are also rewired such that disease-related genes are differentially expressed compared to controls.
In the other study, another team of investigators observed in mouse models and in a three-dimensional human neuronal cell culture that a Herpseviridae infection could seed amyloid-β plaques.
“These two papers add to a weight of evidence that viruses—and pathogens in general—must now be seriously considered as causal agents in Alzheimer’s disease,” Chris Carter, who studies the genetics and epidemiology of Alzheimer’s and other neurological disorders at Polygenic Pathways in the U.K., tells The Scientist.
Over three decades, there have been accumulating data from human studies suggesting that certain microbes, namely, viruses bacteria and fungi, may trigger or promote Alzheimer’s pathology in the aging brain.
The Mount Sinai group initially set out to mine their RNA and DNA sequencing data from Alzheimer’s brain samples for drug targets. Then they found these viral sequences that were difficult to ignore. “I recently gave a talk that I titled, ‘I went looking for drugs but all I found was these viruses,’” study coauthor Joel Dudley, a genomics researcher at the Icahn School of Medicine at Mount Sinai, tells The Scientist.
In their study of elderly human brains, Dudley and the team from Mount Sinai sequenced more than 1,400 post-mortem brain samples, finding the first evidence that human herpes viruses 6A (HHV-6A) and 7 (HHV-7) are in greater abundance in regions of the brain including the superior temporal gyrus, anterior prefrontal cortex, and dorsolateral prefrontal cortex.
Using RNA and DNA sequencing data, the team computationally generated regulatory network models that implicated the presence these viruses in altering the activity of genes linked to Alzheimer’s risk.
The researchers turned to one of the microRNAs, miR-155, found in their analysis to be suppressed by HHV-6A in the human samples, to see what the functional consequence is of this interaction. They homed in on miR-155 because it was a novel microRNA and because it had been previously linked to herpes viruses. When they knocked out the gene for miR-155 in a mouse model of Alzheimer’s disease, the animals’ brains had larger amyloid plaques and higher levels of amyloid-β compared to the mouse model with a wildtype MIR155 gene.
“Conceivably, the viral proteins are acting as transcription factors that control expression of Alzheimer’s risk genes,” co-author Sam Gandy, a professor of neurology who specializes in Alzheimer’s disease at Mount Sinai, writes in an email to The Scientist. “Perhaps this viral dysregulation of Alzheimer’s genes that we see promotes the Alzheimer’s pathology of amyloid beta aggregation, inflammation and tau tangles,” he says.
The results, published today (June 21) in Neuron, could pave the way to new intervention strategies. “If established that these viruses indeed play a role in the development of Alzheimer’s, retroviral agents should be tested as a potential therapy,” says Dudley.
In the other study, available as a preprint on the Cell website and in Neuron July 11, Rudolph Tanzi and Robert Moir, both researchers at Harvard Medical School and Massachusetts General Hospital, and their colleagues tested how amyloid-β in the brain—which these labs previously found to be an antimicrobial—reacts to herpes simplex virus 1 (HSV1), HHV6A, and HHV6B. These strains all tend to integrate into the genomes of neurons. They found that in a culture of human neuronal cells, amyloid-β could prevent HSV1 infection and can bind and aggregate the HSV1 and HHV6 viruses. Mice infected with HSV1—which can cause encephalitis—that also had genetically elevated amyloid-β expression were protected against encephalitis, but also had increased amyloid deposits.
“These studies further add to the steadily increasing number of papers that support a microbial role in Alzheimer’s disease,” Ruth Itzhaki, a molecular neurobiologist at the University of Manchester in the U.K. who studies the link between viruses and the development of Alzheimer’s disease, writes in an email to The Scientist.
A recent epidemiology study adds real-world credence to the microbial link to Alzheimer’s. A population study in Taiwan examined more than 33,000 individuals and found that those with a herpes simplex virus infection had a 2.5-fold greater risk of developing Alzheimer’s disease. The study authors found that in those people treated with anti-herpes medications, the 2.5-fold risk dropped back down to baseline.
“The conclusion you can draw is that the antiherpes medication reduced the risk of Alzheimer’s by keeping the herpes infection in check,” says Moir.
Itzhaki agrees. This study and two others, also from Taiwan, appear to link HSV1 causally to Alzheimer’s disease, she writes. “Despite various shortcomings, these Taiwan studies are the essential first steps to a proof that a microbe could be the cause of a non-infectious disease, in this case, Alzheimer’s.” Itzhaki and a colleague wrote about these studies recently in a commentary, which aimed to interpret the “important and surprising Taiwan data” on the effectiveness of the antiviral treatment, Itzhaki tells The Scientist.
Carter cautions that the new reports should not be interpreted to mean that there is likely a single, unique Alzheimer’s pathogen, if there is one at all. “These data suggest that multiple pathogens, and not just these viruses, likely contribute to Alzheimer’s disease. It is also likely that the pathogens may vary between Alzheimer’s patients.”
The Mount Sinai team will now be verifying whether HHV6 and HHV7 are actually integrated into the genomes of Alzheimer’s patients’ brains and testing for the presence of HHV6 and HHV7 in the bloodstream and central nervous system of Alzheimer’s patients. They would like to do a study comparing living patients and controls to see if the link they observed between the viruses’ presence and changes in gene regulation related to Alzheimer’s holds up.
Tanzi’s and Moir’s labs are focusing on the role of the brain microbiome in Alzheimer’s disease. Comparing the brains of older and younger individuals, including those with Alzheimer’s, their preliminary evidence shows that the brain microbiome—which contains hundreds of bacterial and fungal species—is shifted and linked to pro-inflammatory activity. “It’s analogous to what happens with the gut microbiome in individuals with irritable bowel syndrome,” says Moir. “Our model right now is that it’s not just a single microbe, but a disturbance in the brain microbiome that can lead to Alzheimer’s disease.”
https://www.the-scientist.com/news-opinion/herpes-viruses-implicated-in-alzheimer-s-disease-64246
-----
Do microbes trigger Alzheimer’s Disease?
The once fringe idea is gaining traction among the scientific community.
n late 2011, Drexel University dermatology professor Herbert Allen was astounded to read a new research paper documenting the presence of long, corkscrew-shape bacteria called spirochetes in postmortem brains of patients with Alzheimer’s disease.1 Combing data from published reports, the International Alzheimer Research Center’s Judith Miklossy and colleagues had found evidence of spirochetes in 451 of 495 Alzheimer’s brains. In 25 percent of cases, researchers had identified the spirochete as Borrelia burgdorferi, a causative agent of Lyme disease. Control brains did not contain the spirochetes.
The study made Allen think back to 40 years earlier, when he was an intern at Johns Hopkins University and had treated a patient diagnosed with neurosyphilis, a neurological syndrome that included dementia and resulted from the invasion of the syphilis spirochete into the brain. “The parallel between Lyme disease and syphilis had me intrigued,” he says.
Allen had recently proposed a novel role for biofilms—colonies of bacteria that adhere to surfaces and are largely resistant to immune attack or antibiotics—in eczema. He suggested that because biofilms block skin ducts and trigger innate immune responses, they may cause the stubborn skin condition. Allen knew of recent work showing that Lyme spirochetes form biofilms, which led him to wonder if biofilms might also play a role in Alzheimer’s disease. When Allen stained for biofilms in brains from deceased Alzheimer’s patients, he found them in the same hippocampal locations as amyloid plaques. Toll-like receptor 2 (TLR2), a key player in innate immunity, was also present in the same region of the Alzheimer’s brains but not in the controls. He hypothesizes that TLR2 is activated by the presence of bacteria, but is locked out by the biofilm and damages the surrounding tissue instead.
Spirochetes, common members of the oral microbiome, belong to a small set of microbes that cross the blood-brain barrier when they’re circulating in the blood, as they are during active Lyme infections or after oral surgery. However, the bacteria are so slow to divide that it can take decades to grow a biofilm. This time line is consistent with Alzheimer’s being a disease of old age, Allen reasons, and is corroborated by syphilis cases in which the neuroinvasive effects of spirochetes might appear as long as 50 years after primary infection.
Allen’s work contributes to the revival of a long-standing hypothesis concerning the development of Alzheimer’s. For 30 years, a handful of researchers have been pursuing the idea that pathogenic microbes may serve as triggers for the disease’s neuropathology. Most came across the connection serendipitously, as Allen did, and some have made it their life’s work, in spite of scathing criticism and related challenges in attracting funding and publishing results.
“There have been all these observations over time,” says Miklossy. Although she says she’s been dismissed as an “idiot” and denied funding, she continues to pursue spirochetes as an instigating factor in Alzheimer’s disease. “I’m a physician who believes in the Hippocratic Oath,” she says. “We have to do everything we can.”
And the Alzheimer’s field seems primed for a fresh look at a theory that might account for the disease’s pathogenesis. Researchers still cannot say with confidence which features of the disease, such as neuroinflammation, tau tangles, and amyloid plaques, are involved in disease progression and thus would make effective targets for treatment. So far, most drugs that have made it to clinical testing have targeted the amyloid-β peptide, the main component of the amyloid plaques that characterize Alzheimer’s brains. The idea is that a build-up of amyloid-β causes the neuropathology and that removing amyloid-β—by decreasing its production, impeding aggregation, or aiding removal of the molecule from the brain—will improve, or at least stall, symptoms of dementia. But so far, researchers have come up empty-handed.
Last November, for example, a Phase 3 trial of Eli Lilly’s amyloid-targeting antibody solanezumab revealed no improvement in patients in early stages of the disease. This costly and crushing failure was followed up a few months later with another, when Merck halted its clinical trial of the small-molecule drug verubecestat, which blocks the enzyme that yields amyloid-β, in patients with mild to moderate disease. A trial using verubecestat in the earliest diagnosable stage of the disease is still underway.
And these are just the latest in a string of experimental drugs for Alzheimer’s disease that have failed to show any benefit in clinical trials. Some blame the trials themselves for these high-profile flops. “The quality of the clinical trials has been low,” says John Hardy, a molecular neuroscientist at the University College London, pointing out that a couple of the drugs didn’t even make it into the brain. But other researchers question the underlying theory.
In light of continued failures to develop effective drugs, some researchers, such as Harvard neurobiologist Rudolph Tanzi, think it’s high time that more effort and funding go into alternative theories of the disease. “Any hypothesis about Alzheimer’s disease must include amyloid plaques, tangles, inflammation—and, I believe, infection.”
A history of microbial links
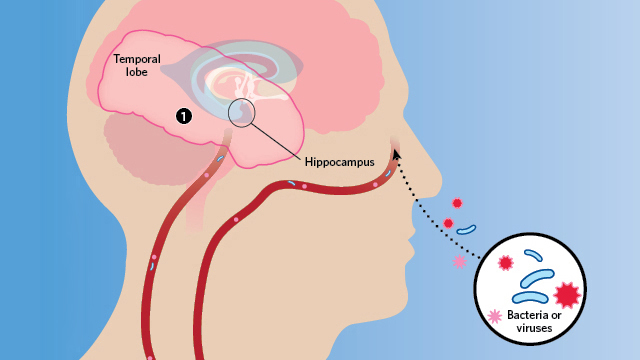
Emerging evidence links bacterial or viral infection with the neuropathology of Alzheimer’s.
Herpes simplex virus type 1 (HSV1) can acutely infect the brain and cause a rare but very serious encephalitis. In the late 1980s, University of Manchester molecular virologist Ruth Itzhaki noticed that the areas of the brain affected in HSV1 patients were the same as those damaged in patients with Alzheimer’s disease. Knowing that herpes can lie latent in the body for long periods of time, she began to wonder if there was a causal connection between the infection and the neurodegenerative disorder.
Itzhaki began looking for HSV1 DNA in the brains of Alzheimer’s patients—and found it. But the viral DNA also turned up in the brains of age-matched controls. Using PCR, still a new technique at the time, was fraught with difficulty, and Itzhaki’s findings were challenged as resulting from contamination. Itzhaki repeated her work with great care and consistently found that two-thirds to three-quarters of elderly people harbor HSV1 in their brains, whether they had Alzheimer’s or not. So she searched for a genetic difference that might explain why only some HSV1-infected individuals develop dementia. Finally, in 1997, she reported that having both HSV1 in the brain and the apolipoprotein E gene variant APOE4 accounted for 60 percent of the Alzheimer’s cases she considered—much higher than either factor alone.5 But most Alzheimer’s researchers still dismissed her work. Itzhaki says her detractors have been set in their ways—and perhaps too wedded to scenarios involving plaques and tangles. “They don’t know anything about viruses,” she says, especially the fact that herpes can linger in the body and brain. “If we say the virus causes this, they imagine the scenario is fast. It’s incredibly naive.”
Around the same time, neuropathologist Miklossy, then at the University of Lausanne in Switzerland, was detailing the brain damage caused by spirochetes—both in neurosyphilis and neuroborrelia, a syndrome caused by Lyme bacteria. She happened upon a head trauma case with evidence of bacterial invasion and plaque formation, and turned her attention to Alzheimer’s. She isolated spirochetes from brain tissue in 14 Alzheimer’s patients but detected none in 13 age-matched controls. In addition, monoclonal antibodies that target the amyloid precursor protein (APP)—which, when cleaved, forms amyloid-β—cross-reacted with the spirochete species found, suggesting the bacteria might be the source of the protein.
Although Miklossy says she received some positive reactions to her findings when she published them in 1993, she, like Itzhaki, also faced her fair share of skepticism. The critiques included comments that the work was “foolish, unorthodox, and crazy,” she adds.
Meanwhile, in the U.S., a third line of evidence linking Alzheimer’s to microbial infection began to emerge. While serving on a fraud investigation committee, Alan Hudson, a microbiologist then at MCP-Hahnemann School of Medicine in Philadelphia, met Brian Balin, who studied neuropathological processes at the Philadelphia College of Osteopathic Medicine. Soon, Balin began to send Hudson Alzheimer’s brain tissue to test for intracellular bacteria in the Chlamydia genus. Some samples tested positive for C. pneumoniae: specifically, the bacteria resided in microglia and astrocytes in regions of the brain associated with Alzheimer’s neuropathology, such as the hippocampus and other limbic system areas. Hudson had a second technician repeat the tests before he called Balin to unblind the samples. The negatives were from control brains; the positives all had advanced Alzheimer’s disease. “We were floored,” Hudson says.
The paper that Balin, Hudson, and colleagues wrote to announce the findings received worldwide press coverage, says Hudson, now professor emeritus at Wayne State University School of Medicine. But when the authors went to the Alzheimer’s disease meeting, he says, “nobody talked to us.”
Thus, as early as the 1990s, three laboratories in different countries, each studying different organisms, had each implicated human pathogens in the etiology of Alzheimer’s disease. But the suggestion that Alzheimer’s might have some microbial infection component was still well outside of the theoretical mainstream.
New century, new mechanisms
Last year, Itzhaki, Miklossy, Hudson, and Balin, along with 29 other scientists, published a review in the Journal of Alzheimer’s Disease to lay out the evidence implicating a causal role for microbes in the disease. The paper opens with a plea: “We are researchers and clinicians working on Alzheimer’s disease . . . and we write to express our concern that one particular aspect of the disease has been neglected.”
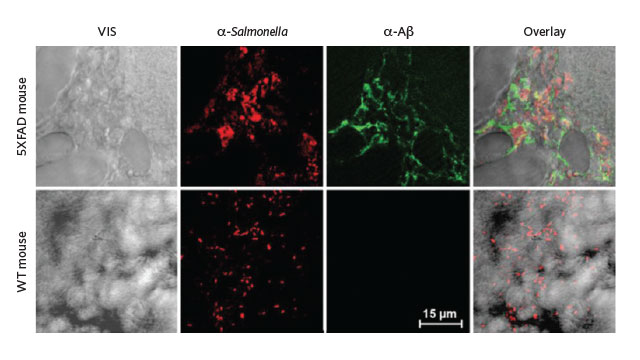
Transgenic mice (top row) whose brains were injected with Salmonella expressed high levels
of amyloid-β in those same regions 48 hours later.
George Perry, editor of the journal and an Alzheimer’s researcher at the University of Texas at San Antonio, not only agreed to publish the article, he signed on as an author too. “The Journal of Alzheimer’s Disease promotes all sorts of different ideas,” he says. “We don’t care about popularity.”
And, slowly but surely, Alzheimer’s researchers finally seem to be giving the pathogen hypothesis a good, hard look. Harvard’s Tanzi, one of the newer microbial theorists, has been a prominent figure in the Alzheimer’s field for decades. He contributed to the 1987 discovery of APP, the first Alzheimer’s disease gene. Recently, Tanzi and his colleagues showed that amyloid-β inhibits the in vitro growth of pathogenic bacteria, including Candida albicans, E. coli, and Staphylococcus aureus, suggesting the Alzheimer’s-linked peptide was acting as an antimicrobial. Tanzi’s working hypothesis is that microbes trigger an innate immune response, in which amyloid-β plays a key role. The peptide surrounds the site of infection to shield healthy tissue from the invaders. Too much clumping, however, can cause problems of its own—the very processes by which plaques trigger neuronal death.
A subsequent study by Tanzi’s group found that amyloid-β binds to microbes and links together with more amyloid-β to entrap the invaders and keep them from interacting with host cells. Indeed, in a transgenic mouse model of Alzheimer’s disease, Salmonella infection seeded amyloid plaques in the brain. “The plaques are generated in the hippocampus and temporal cortex—the regions most susceptible to blood-brain barrier breach,” Tanzi says, suggesting that those areas are where pathogens would first gain entry. “It makes perfect sense to me.”
Tanzi is well aware of the work by Miklossy and others and the criticism that they’ve received. Expecting to get their own dose of criticism, Tanzi says, “we wanted to do everything right, do every control. We spent eight years on this paper.” But to his surprise, the backlash didn’t come. “To our delight, the field looked at what we did,” he says—a sign, perhaps, that the Alzheimer’s research community is finally ready to consider the microbe theory.
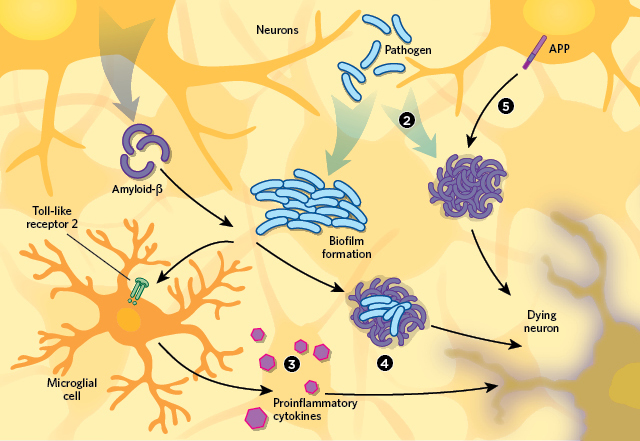
There are several hypotheses to explain the apparent connection between infection and
Alzheimer’s neurodegeneration pathology.
Proponents of this idea still face skepticism, however. Elaine Bearer, a molecular neurobiologist at the University of New Mexico Health Sciences Center, received mixed responses when she began publishing and presenting her work linking HSV1 to Alzheimer’s neuropathology. As is a familiar story by now, Bearer had stumbled onto the microbe theory serendipitously.
Her main research interest is how molecular motors pick up cargo in the giant squid axon, and she uses HSV1 as experimental cargo because it’s known to travel in both directions along axonal transport routes. During infection, HSV1 travels from sensory nerve endings to nerve cell bodies where the virus can hole up. When activated, HSV1 travels back out to synapses, reinfects epithelial cells, and voile—cold-sore recurrence.
In 2006, Bearer found that HSV1 uses APP to attach to axonal transport machinery. And as a result, HSV1 redistributes APP within the neuron, she says. That means APP can pile up in ways that don’t happen in uninfected cells. More recently, Bearer showed that “the virus does something to APP,” she says. “In epithelial cells, it induces a 25-fold increase in the protein,” suggesting synthesis of the protein also responds to infection.
Bearer has also produced evidence that HSV1 is trapped in amyloid plaques in human brains. (She has presented this work, but not published it.) This mirrors Tanzi’s findings of Salmonella within amyloid plaques in an animal model, and those of Allen, who found bacterial biofilms colocalized with amyloid-β in human brain tissue.
Despite the increase in evidence supporting the microbial theory of Alzheimer’s disease, however, funding for such research remains difficult to procure. And scientists working in this area also continue to face skepticism from the Alzheimer’s research community. University College London’s Hardy, squarely in the amyloid hypothesis camp, is aware of the work of Itzhaki, Tanzi, and some of the others, but he says he’s still “not convinced.”
Hardy’s main objections are twofold: the idea that microbes cause Alzheimer’s neuropathology doesn’t fully explain the hereditary aspects of the disease, and it doesn’t explain the characteristic anatomical distribution of plaques and tangles in diseased brains. He thinks distribution would be more widespread in the brain with blood-borne disease. “It just doesn’t ring right,” he says. “It doesn’t fit the epidemiology, the neuropathology, or the genetics.” To get him to change his tune, Hardy says, he’d need to see more experimental evidence “to show some element of cause and effect: infect mice, infect primates, and show disease.”
The microbe theorists freely admit that their proposed microbial triggers are not the only cause of Alzheimer’s disease. In Itzhaki’s case, some 40 percent of cases are not explained by HSV1 infection. Of course, the idea that Alzheimer’s might be linked to infection isn’t limited to any one pathogen; the hypothesis is simply that, following infection, certain pathogens gain access to brain, where immune responses result in the accumulation of amyloid-β, leading to plaque formation. In the meantime, with Alzheimer’s patients representing a huge unmet medical need, and experimental drugs often failing in late-stage trials, even Hardy admits that there are more questions than answers at this point in terms of the causative factors in Alzheimer’s. “The pathology is a mess. The brain has been diseased for a long time by the time we see it,” Hardy says. “We’re looking at the end product and trying to determine how it got that way.”
Perry agrees: “Most of the resources in this field are spent on a few biomarkers. All the evidence shows that amyloid is important. But causality and centrality are two different things.”
https://www.the-scientist.com/features/do-microbes-trigger-alzheimers-disease-30999
Last edited: