S.J.B.
Bluelight Crew
- Joined
- Jan 22, 2011
- Messages
- 6,886
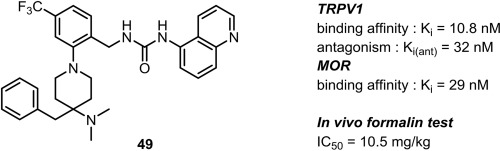
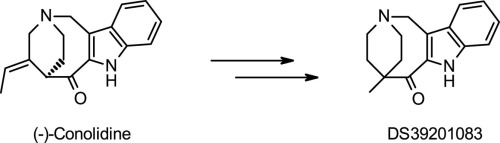
Discovery of conolidine derivative DS39201083 as a potent novel analgesic without mu opioid agonist activity
Corresponding author: Tsuyoshi Arita (R&D Division, Daiichi Sankyo Co., Ltd., Tokyo, Japan)
Bioorganic & Medicinal Chemistry Letters 2019, Volume 29, Issue 15, Pages 1938-1942
Published online May 21st, 2019
https://doi.org/10.1016/j.bmcl.2019.05.045
Corresponding author: Tsuyoshi Arita (R&D Division, Daiichi Sankyo Co., Ltd., Tokyo, Japan)
Bioorganic & Medicinal Chemistry Letters 2019, Volume 29, Issue 15, Pages 1938-1942
Published online May 21st, 2019
https://doi.org/10.1016/j.bmcl.2019.05.045
We discovered a novel compound, 5-methyl-1,4,5,7-tetrahydro-2,5-ethanoazocino[4,3-b]indol-6(3H)-one sulfuric acid salt (DS39201083), which was formed by derivatization of a natural product, conolidine. DS39201083 had a unique bicyclic skeleton and was a more potent analgesic than conolidine, as revealed in the acetic acid-induced writhing test and formalin test in ddY mice. The compound showed no agonist activity at the mu opioid receptor.


")