-
N&PD Moderators: Skorpio | thegreenhand
-
Neuroscience & Pharmacology Discussion Welcome Guest
Posting Rules Bluelight Rules Recent Journal Articles Chemistry Mega-Thread FREE Chemistry Databases! Self-Education Guide
You are using an out of date browser. It may not display this or other websites correctly.
You should upgrade or use an alternative browser.
You should upgrade or use an alternative browser.
⫸STICKY⫷ The N&PD Recent Journal ARTICLE Club
- Thread starter nuke
- Start date
- Joined
- Jul 2, 2008
- Messages
- 9,131
So looks like there is some actual proof about the harmful effects of nicotine itself... Is this some unspecific effect of nicotine or is it a direct result of binding to nicotinic receptors? Another nicotine agonist that is found in nature is the arecoline in Betel nut, but that compound is also a muscarine agonist to some extent.
In mice. I wouldn't advocate pregnant women smoking, or anyone for that matter, but back in the 50's just about everyone smoked. And that was for hundreds of years, since the 1500's, if I recall right. Now, understanding that there might be more unhealthy additives today as opposed to back then, I still think that the pure plant isn't quite neurologically toxic. For example, so many of the groundbreaking philosophers in The Enlightenment were smokers, which itself really began in the 1500's. I don't quite buy the "well they would have been even better without it" theory.
As far as I'm aware, all tobacco has specific nitrosamines that makes the plant carcinogenic (without additives). But in terms of causing dementia/Alzheimers, I remain skeptical.
Since smoking once upon a time was everywhere, perhaps people sitting in their dens/man caves of then would think better while smoking there, but run a little deficit otherwise, which might explain smokers having compromised cognition in studies wherein they weren't smoking. It's like caffeine. It still works to an extent even if you're addicted, yet sets you at a disadvantage without it.
polymath
Bluelight Crew
- Joined
- Nov 4, 2010
- Messages
- 1,884
In mice. I wouldn't advocate pregnant women smoking, or anyone for that matter, but back in the 50's just about everyone smoked. And that was for hundreds of years, since the 1500's, if I recall right. Now, understanding that there might be more unhealthy additives today as opposed to back then, I still think that the pure plant isn't quite neurologically toxic. For example, so many of the groundbreaking philosophers in The Enlightenment were smokers, which itself really began in the 1500's. I don't quite buy the "well they would have been even better without it" theory.
Yeah, I think that in the 18th century there were many places in the world where almost no one smoked tobacco, but things like calculus and classical mechanics were still developed first in Europe where smoking was common. It may be that nicotine exposure just makes it more likely for one to produce ADHD offspring but doesn't affect the likelyhood of genius children being born.
In the future, it will be possible to do gene edits that improve intelligence - for instance the deletion of the gene that codes the alpha5 subunit of GABA-A receptor improves memory and learning in lab rats without causing any obvious harmful effects. But of course, before doing such gene deletions to people it would be safest to first try to find some rare individual who happens to have such a mutation by random (in nuclear disaster sites there are probably many people with unusual mutations) and see whether they suffer from any harmful side effects. Just writing this because of the recent news about gene edited babies in China...
- Joined
- Jul 2, 2008
- Messages
- 9,131
Wow, didn't know about that in China. Not terribly surprised that they would do so. What I do know is that clones die much quicker than naturally-conceived animals, three times as fast. Gene editing is a huge moral can of worms, for one.
polymath
Bluelight Crew
- Joined
- Nov 4, 2010
- Messages
- 1,884
There are now some methods to change the genes of an adult person, too. I think some "biohacker" already tested it on himself. It's likely that there will be plenty of willing guinea pigs for that kind of thing, including athletes who want to increase their physical performance.
sekio
Bluelight Crew
- Joined
- Sep 14, 2009
- Messages
- 21,994
How about a retrovirus that patches the Vitamin C synthesis enzyme (L-gulonolactone oxidase) back to working order. That'd be cool, being able to produce multiple grams of VitC a day with no need for supplementation.
Or make a baby that produces green fluorescent protein in his skin. Fluorescent baby. Or make the sclera of the eyes produce a blue fluorescent protien and you can make a race of Fremen from Dune.
Or make a baby that produces green fluorescent protein in his skin. Fluorescent baby. Or make the sclera of the eyes produce a blue fluorescent protien and you can make a race of Fremen from Dune.
polymath
Bluelight Crew
- Joined
- Nov 4, 2010
- Messages
- 1,884
An even better "sci-fi" gene edit would be to add the tardigrade radiation tolerance gene to human DNA, so that people could withstand long-term space travel...
DNA Protection Protein, a Novel Mechanism of Radiation Tolerance: Lessons from Tardigrades
DNA Protection Protein, a Novel Mechanism of Radiation Tolerance: Lessons from Tardigrades
Genomic DNA stores all genetic information and is indispensable for maintenance of normal cellular activity and propagation. Radiation causes severe DNA lesions, including double-strand breaks, and leads to genome instability and even lethality. Regardless of the toxicity of radiation, some organisms exhibit extraordinary tolerance against radiation. These organisms are supposed to possess special mechanisms to mitigate radiation-induced DNA damages. Extensive study using radiotolerant bacteria suggested that effective protection of proteins and enhanced DNA repair system play important roles in tolerability against high-dose radiation. Recent studies using an extremotolerant animal, the tardigrade, provides new evidence that a tardigrade-unique DNA-associating protein, termed Dsup, suppresses the occurrence of DNA breaks by radiation in human-cultured cells. In this review, we provide a brief summary of the current knowledge on extremely radiotolerant animals, and present novel insights from the tardigrade research, which expand our understanding on molecular mechanism of exceptional radio-tolerability.
polymath
Bluelight Crew
- Joined
- Nov 4, 2010
- Messages
- 1,884
Here's a new article about functionally selective kappa agonists (apparently there's two different secondary messengers that can be activated by binding).
Structurally Related Kappa Opioid Receptor Agonists with Substantial Differential Signaling Bias: Neuroendocrine and Behavioral Effects in C57BL6 Mice Int J Neuropsychopharmacol. 2018 Sep; 21(9): 847?857.
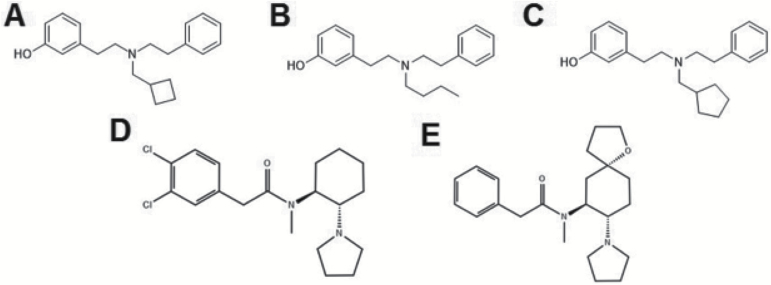
I'm not sure about which kind of functional selectivity is behind the salvia-type hallucinogenic effects, but the compound B (BPHA) would seem to be relatively easy to produce synthetically. However, that's the one compound that caused least motor incoordination.
Is there any SAR available for those phenylacetic amide compounds D and E?
Structurally Related Kappa Opioid Receptor Agonists with Substantial Differential Signaling Bias: Neuroendocrine and Behavioral Effects in C57BL6 Mice Int J Neuropsychopharmacol. 2018 Sep; 21(9): 847?857.
Abstract
Background
The kappa opioid receptor system has been revealed as a potential pharmacotherapeutic target for the treatment of addictions to substances of abuse. Kappa opioid receptor agonists have been shown to block the rewarding and dopamine-releasing effects of psychostimulants. Recent investigations have profiled the in vivo effects of compounds biased towards G-protein-mediated signaling, with less potent arrestin-mediated signaling. The compounds studied here derive from a series of trialkylamines: N-substituted-N- phenylethyl-N-3-hydroxyphenylethyl-amine, with N-substituents including n-butyl (BPHA), methylcyclobutyl (MCBPHA), and methylcyclopentyl (MCPPHA).
Methods
BPHA, MCBPHA, and MCPPHA were characterized in vitro in a kappa opioid receptor-expressing cell line in binding assays and functional assays. We also tested the compounds in C57BL6 mice, assaying incoordination with rotarod, as well as circulating levels of the neuroendocrine kappa opioid receptor biomarker, prolactin.
Results
BPHA, MCBPHA, and MCPPHA showed full kappa opioid receptor agonism for G-protein coupling compared with the reference compound U69,593. BPHA showed no measurable β-arrestin-2 recruitment, indicating that it is extremely G-protein biased. MCBPHA and MCPPHA, however, showed submaximal efficacy for recruiting β-arrestin-2. Studies in C57BL6 mice reveal that all compounds stimulate release of prolactin, consistent with dependence on G-protein signaling. MCBPHA and MCPPHA result in rotarod incoordination, whereas BPHA does not, consistent with the reported requirement of intact kappa opioid receptor/β-arrestin-2 mediated coupling for kappa opioid receptor agonist-induced rotarod incoordination.
Conclusions
BPHA, MCBPHA, and MCPPHA are thus novel differentially G-protein-biased kappa opioid receptor agonists. They can be used to investigate how signaling pathways mediate kappa opioid receptor effects in vitro and in vivo and to explore the effects of candidate kappa opioid receptor-targeted pharmacotherapeutics.
I'm not sure about which kind of functional selectivity is behind the salvia-type hallucinogenic effects, but the compound B (BPHA) would seem to be relatively easy to produce synthetically. However, that's the one compound that caused least motor incoordination.
Is there any SAR available for those phenylacetic amide compounds D and E?
polymath
Bluelight Crew
- Joined
- Nov 4, 2010
- Messages
- 1,884
It was already known in the 1990s that neuropeptide FF receptor blockers prevent opioid tolerance, but now there's a compound with both effects in one.
A bifunctional-biased mu-opioid agonist-neuropeptide FF receptor antagonist as analgesic with improved acute and chronic side effects. Pain. 2018 Sep;159(9):1705-1718. doi: 10.1097/j.pain.0000000000001262.
A bifunctional-biased mu-opioid agonist-neuropeptide FF receptor antagonist as analgesic with improved acute and chronic side effects. Pain. 2018 Sep;159(9):1705-1718. doi: 10.1097/j.pain.0000000000001262.
Abstract
Opioid analgesics, such as morphine, oxycodone, and fentanyl, are the cornerstones for treating moderate to severe pain. However, on chronic administration, their efficiency is limited by prominent side effects such as analgesic tolerance and dependence liability. Neuropeptide FF (NPFF) and its receptors (NPFF1R and NPFF2R) are recognized as an important pronociceptive system involved in opioid-induced hyperalgesia and analgesic tolerance. In this article, we report the design of multitarget peptidomimetic compounds that show high-affinity binding to the mu-opioid receptor (MOPr) and NPFFRs. In vitro characterization of these compounds led to identification of KGFF03 and KGFF09 as G-protein-biased MOPr agonists with full agonist or antagonist activity at NPFFRs, respectively. In agreement with their biased MOPr agonism, KGFF03/09 showed reduced respiratory depression in mice, as compared to the unbiased parent opioid agonist KGOP01. Chronic subcutaneous administration of KGOP01 and KGFF03 in mice rapidly induced hyperalgesia and analgesic tolerance, effects that were not observed on chronic treatment with KGFF09. This favorable profile was further confirmed in a model of persistent inflammatory pain. In addition, we showed that KGFF09 induced less physical dependence compared with KGOP01 and KGFF03. Altogether, our data establish that combining, within a single molecule, the G-protein-biased MOPr agonism and NPFFR antagonism have beneficial effects on both acute and chronic side effects of conventional opioid analgesics. This strategy can lead to the development of novel and potent antinociceptive drugs with limited side effects on acute and chronic administration.
sekio
Bluelight Crew
- Joined
- Sep 14, 2009
- Messages
- 21,994
Sulforaphane Inhibited the Nociceptive Responses, Anxiety- and Depressive-Like Behaviors Associated With Neuropathic Pain and Improved the Anti-allodynic Effects of Morphine in Mice
Pablo Ferreira-Chamorro, Alejandro Redondo, Gabriela Riego, Sergi Le?nez and Olga Pol
https://www.frontiersin.org/articles/10.3389/fphar.2018.01332/full
Our results showed that the repeated administration of SFN besides inhibiting nociceptive responses induced by sciatic nerve injury also diminished the anxiety- and depressive-like behaviors associated with persistent neuropathic pain. Moreover, SFN treatment normalized oxidative stress by inducing Nrf2/HO-1 signaling, reduced microglial activation and JNK, ERK1/2, p-38 phosphorylation induced by sciatic nerve injury in the spinal cord and/or hippocampus and prefrontal cortex. Interestingly, treatment with SFN also potentiated the antiallodynic effects of morphine in sciatic nerve-injured mice by regularizing the down regulation of MOR in the spinal cord and/or hippocampus. This study suggested that treatment with SFN might be an interesting approach for the management of persistent neuropathic pain and comorbidities associated as well as to improve the analgesic actions of morphine.
Eat your broccoli, folks.
Pablo Ferreira-Chamorro, Alejandro Redondo, Gabriela Riego, Sergi Le?nez and Olga Pol
https://www.frontiersin.org/articles/10.3389/fphar.2018.01332/full
Our results showed that the repeated administration of SFN besides inhibiting nociceptive responses induced by sciatic nerve injury also diminished the anxiety- and depressive-like behaviors associated with persistent neuropathic pain. Moreover, SFN treatment normalized oxidative stress by inducing Nrf2/HO-1 signaling, reduced microglial activation and JNK, ERK1/2, p-38 phosphorylation induced by sciatic nerve injury in the spinal cord and/or hippocampus and prefrontal cortex. Interestingly, treatment with SFN also potentiated the antiallodynic effects of morphine in sciatic nerve-injured mice by regularizing the down regulation of MOR in the spinal cord and/or hippocampus. This study suggested that treatment with SFN might be an interesting approach for the management of persistent neuropathic pain and comorbidities associated as well as to improve the analgesic actions of morphine.
Eat your broccoli, folks.
S.J.B.
Bluelight Crew
- Joined
- Jan 22, 2011
- Messages
- 6,886
Assessment of the Abuse Potential of Cebranopadol in Nondependent Recreational Opioid Users: A Phase 1 Randomized Controlled Study
Corresponding author: Marie-Henriette Eerdekens (Clinical Science Pain, Grunenthal GmbH, Aachen, Germany)
Journal of Clinical Psychopharmacology 2019, Volume 39, Issue 1, Pages 46-56
Published online December 7th, 2018
https://doi.org/10.1097/jcp.0000000000000995
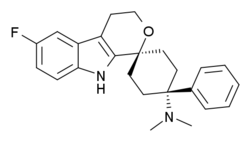
Corresponding author: Marie-Henriette Eerdekens (Clinical Science Pain, Grunenthal GmbH, Aachen, Germany)
Journal of Clinical Psychopharmacology 2019, Volume 39, Issue 1, Pages 46-56
Published online December 7th, 2018
https://doi.org/10.1097/jcp.0000000000000995
Cebranopadol:Background: Cebranopadol is a nociceptin/orphanin FQ peptide/opioid receptor agonist with central antinociceptive activity. We hypothesize that this novel mechanism of action may lead to a lower risk of abuse compared with pure μ-opioid peptide receptor agonists.
Methods: We conducted a single-dose, nested-randomized, double-blind crossover study in nondependent recreational opioid users to evaluate the abuse potential of single doses of cebranopadol relative to hydromorphone immediate release and placebo. The study consisted of a qualification phase and a 7-period treatment phase (cebranopadol 200, 400, and 800 μg; hydromorphone 8 and 16 mg; and 2 placebos). The primary end point was the peak effect of drug liking at this moment, measured by visual analog scale (VAS). Various secondary end points (eg, VAS rating for good drug effects, high, bad drug effects, take drug again, drug similarity, and pupillometry) were also investigated.
Results: Forty-two subjects completed the study. Cebranopadol 200 and 400 μg did not differentiate from placebo on the abuse potential assessments and generated smaller responses than hydromorphone. Responses observed with cebranopadol 800 μg were similar to hydromorphone 8 mg and smaller than hydromorphone 16 mg. The maximum effect for VAS drug liking at this moment was delayed compared with hydromorphone (3 and 1.5 hours, respectively). Cebranopadol administration was safe; no serious adverse events or study discontinuation due to treatment-emergent adverse events occurred.
Conclusions: These results confirm our hypothesis that cebranopadol, a nociceptin/orphanin FQ peptide/opioid receptor agonist, has lower abuse potential than hydromorphone immediate release, a pure μ-opioid peptide agonist.
Last edited:
polymath
Bluelight Crew
- Joined
- Nov 4, 2010
- Messages
- 1,884
^ That's quite good news, because it's best if people with real pain conditions can't ruin their pharmacotherapy by getting addicted to their medications. Maybe that kind of compounds could also dull psychological pain, e.g. panic attacks or paranoid fears.
S.J.B.
Bluelight Crew
- Joined
- Jan 22, 2011
- Messages
- 6,886
Synergistic interaction between the agonism of cebranopadol at nociceptin/orphanin FQ and classical opioid receptors in the rat spinal nerve ligation model
Corresponding author: Thomas Christoph (Preclinical Drug Development, Grunenthal GmbH, Aachen, Germany)
Pharmacology Research & Perspectives 2018, Volume 6, Issue 6, Page e00444
Published online November 28th, 2018
https://doi.org/10.1002/prp2.444
Could Grunenthal be in the lead in the race towards a "less abusable" painkiller? I wonder how the trials are going for this one.
EDIT: the main Phase 3 trial was terminated while a smaller open-label extension of that trial was completed in 2016. That doesn't seem to bode well, but the FDA has a lot of pressure right now to approve any kind of alternative painkiller, so who knows.
Corresponding author: Thomas Christoph (Preclinical Drug Development, Grunenthal GmbH, Aachen, Germany)
Pharmacology Research & Perspectives 2018, Volume 6, Issue 6, Page e00444
Published online November 28th, 2018
https://doi.org/10.1002/prp2.444
Cebranopadol:Cebranopadol (trans‐6'‐fluoro‐4',9'‐dihydro‐N,N‐dimethyl‐4‐phenyl‐spiro[cyclohexane‐1,1'(3'H)‐pyrano[3,4‐b]indol]‐4‐amine) is a novel analgesic nociceptin/orphanin FQ opioid peptide (NOP) and classical opioid receptor (MOP, DOP, and KOP) agonist with highly efficacious and potent activity in a broad range of rodent models of nociceptive, inflammatory, and neuropathic pain as well as limited opioid‐type side effects such as respiratory depression. This study was designed to explore contribution and interaction of NOP and classical opioid receptor agonist components to cebranopadol analgesia in the rat spinal nerve ligation (SNL) model. Assessing antihypersensitive activity in SNL rats intraperitoneal (IP) administration of cebranopadol resulted in ED50 values of 3.3 and 3.58 μg/kg in two independent experiments. Pretreatment (IP) with J‐113397 (4.64 mg/kg) a selective antagonist for the NOP receptor or naloxone (1 mg/kg), naltrindole (10 mg/kg), or nor‐BNI (10 mg/kg), selective antagonists for MOP, DOP, and KOP receptors, yielded ED50 values of 14.1, 16.9, 17.3, and 15 μg/kg, respectively. This 4‐5 fold rightward shift of the dose‐response curves suggested agonistic contribution of all four receptors to the analgesic activity of cebranopadol. Combined pretreatment with a mixture of the antagonists for the three classical opioid receptors resulted in an 18‐fold potency shift with an ED50 of 65.5 μg/kg. The concept of dose equivalence was used to calculate the expected additive effects of the parent compound for NOP and opioid receptor contribution and to compare them with the observed effects, respectively. This analysis revealed a statistically significant difference between the expected additive and the observed effects suggesting intrinsic synergistic analgesic interaction of the NOP and the classical opioid receptor components of cebranopadol. Together with the observation of limited respiratory depression in rats and humans the synergistic interaction of NOP and classical opioid receptor components in analgesia described in the current study may contribute to the favorable therapeutic index of cebranopadol observed in clinical trials.
Could Grunenthal be in the lead in the race towards a "less abusable" painkiller? I wonder how the trials are going for this one.
EDIT: the main Phase 3 trial was terminated while a smaller open-label extension of that trial was completed in 2016. That doesn't seem to bode well, but the FDA has a lot of pressure right now to approve any kind of alternative painkiller, so who knows.
Last edited:
S.J.B.
Bluelight Crew
- Joined
- Jan 22, 2011
- Messages
- 6,886
5-HT2 receptor binding, functional activity and selectivity in N-benzyltryptamines
Corresponding author: Bruce K. Cassels (Department of Chemistry, Faculty of Sciences, University of Chile, Nunoa, Chile)
PLOS One 2019, Volume 14, Issue 1, Page e0209804
Published online January 10th, 2019
https://doi.org/10.1371/journal.pone.0209804
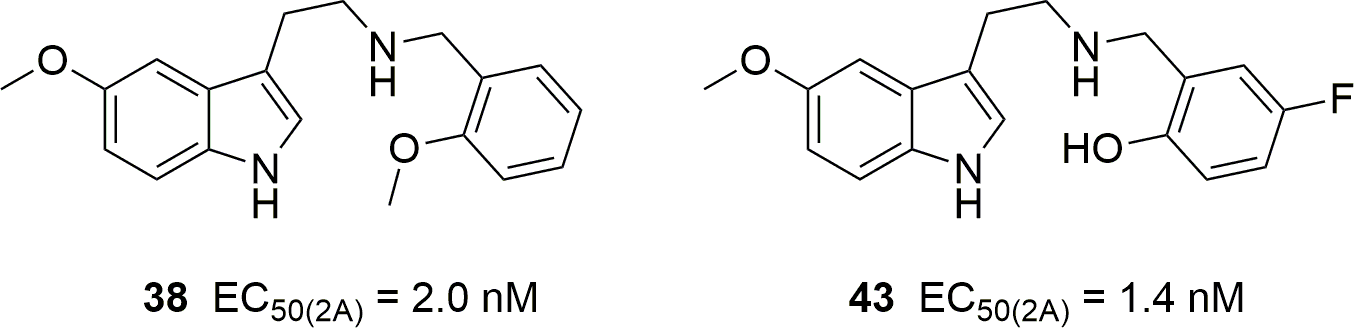
Corresponding author: Bruce K. Cassels (Department of Chemistry, Faculty of Sciences, University of Chile, Nunoa, Chile)
PLOS One 2019, Volume 14, Issue 1, Page e0209804
Published online January 10th, 2019
https://doi.org/10.1371/journal.pone.0209804
The authors suggest that the following two compounds "might be human hallucinogens in the low milligram dose range." The compound on the left had already been studied by Nichols.The last fifteen years have seen the emergence and overflow into the drug scene of "superpotent" N-benzylated phenethylamines belonging to the "NBOMe" series, accompanied by numerous research articles. Although N-benzyl substitution of 5-methoxytryptamine is known to increase its affinity and potency at 5-HT2 receptors associated with psychedelic activity, N-benzylated tryptamines have been studied much less than their phenethylamine analogs. To further our knowledge of the activity of N-benzyltryptamines, we have synthesized a family of tryptamine derivatives and, for comparison, a few 5-methoxytryptamine analogs with many different substitution patterns on the benzyl moiety, and subjected them to in vitro affinity and functional activity assays vs. the human 5-HT2 receptor subtypes. In the binding (radioligand displacement) studies some of these compounds exhibited only modest selectivity for either 5-HT2A or 5-HT2C receptors suggesting that a few of them, with affinities in the 10–100 nanomolar range for 5-HT2A receptors, might presumably be psychedelic. Unexpectedly, their functional (calcium mobilization) assays reflected very different trends. All of these compounds proved to be 5-HT2C receptor full agonists while most of them showed low efficacy at the 5-HT2A subtype. Furthermore, several showed moderate-to-strong preferences for activation of the 5-HT2C subtype at nanomolar concentrations. Thus, although some N-benzyltryptamines might be abuse-liable, others might represent new leads for the development of therapeutics for weight loss, erectile dysfunction, drug abuse, or schizophrenia.
Last edited:
S.J.B.
Bluelight Crew
- Joined
- Jan 22, 2011
- Messages
- 6,886
A bifunctional nociceptin and mu opioid receptor agonist is analgesic without opioid side effects in nonhuman primates
Corresponding authors: Nurulain T. Zaveri (Astraea Therapeutics, Mountain View, United States) and Mei-Chuan Ko (Department of Physiology and Pharmacology, Wake Forest School of Medicine, Winston-Salem, United States)
Science Translational Medicine 2018, Volume 10, Issue 456, Page eaar3483
Published online August 29th, 2018
https://doi.org/10.1126/scitranslmed.aar3483

Corresponding authors: Nurulain T. Zaveri (Astraea Therapeutics, Mountain View, United States) and Mei-Chuan Ko (Department of Physiology and Pharmacology, Wake Forest School of Medicine, Winston-Salem, United States)
Science Translational Medicine 2018, Volume 10, Issue 456, Page eaar3483
Published online August 29th, 2018
https://doi.org/10.1126/scitranslmed.aar3483
AT-121:Misuse of prescription opioids, opioid addiction, and overdose underscore the urgent need for developing addiction-free effective medications for treating severe pain. Mu opioid peptide (MOP) receptor agonists provide very effective pain relief. However, severe side effects limit their use in the clinical setting. Agonists of the nociceptin/orphanin FQ peptide (NOP) receptor have been shown to modulate the antinociceptive and reinforcing effects of MOP agonists. We report the discovery and development of a bifunctional NOP/MOP receptor agonist, AT-121, which has partial agonist activity at both NOP and MOP receptors. AT-121 suppressed oxycodone's reinforcing effects and exerted morphine-like analgesic effects in nonhuman primates. AT-121 treatment did not induce side effects commonly associated with opioids, such as respiratory depression, abuse potential, opioid-induced hyperalgesia, and physical dependence. Our results in nonhuman primates suggest that bifunctional NOP/MOP agonists with the appropriate balance of NOP and MOP agonist activity may provide a dual therapeutic action for safe and effective pain relief and treating prescription opioid abuse.
Last edited:
S.J.B.
Bluelight Crew
- Joined
- Jan 22, 2011
- Messages
- 6,886
Structural Simplification of a Tetrahydroquinoline-Core Peptidomimetic μ-Opioid Receptor (MOR) Agonist/δ-Opioid Receptor (DOR) Antagonist Produces Improved Metabolic Stability
Corresponding author: Henry I. Mosberg (Department of Medicinal Chemistry, College of Pharmacy, University of Michigan, Ann Arbor, United States)
Journal of Medicinal Chemistry 2019, Volume 62, Issue 8, Pages 4142–4157
Published online March 29th, 2019
https://doi.org/10.1021/acs.jmedchem.9b00219

5l: R1 = H, R2 = OEt
5m: R1 = H, R2 = OnPr
Corresponding author: Henry I. Mosberg (Department of Medicinal Chemistry, College of Pharmacy, University of Michigan, Ann Arbor, United States)
Journal of Medicinal Chemistry 2019, Volume 62, Issue 8, Pages 4142–4157
Published online March 29th, 2019
https://doi.org/10.1021/acs.jmedchem.9b00219
We have previously reported a series of μ-opioid receptor (MOR) agonist/δ-opioid receptor (DOR) antagonist ligands to serve as potential nonaddictive opioid analgesics. These ligands have been shown to be active in vivo, do not manifest withdrawal syndromes or reward behavior in conditioned-place preference assays in mice, and do not produce dependence. Although these attributes are promising, these analogues exhibit poor metabolic stability in mouse liver microsomes, likely due to the central tetrahydroquinoline scaffold in this series. As such, a structure–activity relationship (SAR) campaign was pursued to improve their metabolic stability. This resulted in a shift from our original bicyclic tetrahydroquinoline core to a monocyclic benzylic-core system. By eliminating one of the rings in this scaffold and exploring the SAR of this new core, two promising analogues were discovered. These analogues (5l and 5m) had potency and efficacy values at MOR better or comparable to morphine, retained their DOR-antagonist properties, and showed a 10-fold improvement in metabolic stability.
5l: R1 = H, R2 = OEt
5m: R1 = H, R2 = OnPr
Last edited:
S.J.B.
Bluelight Crew
- Joined
- Jan 22, 2011
- Messages
- 6,886
Synthesis and Evaluation of Novel Biased μ-Opioid-Receptor (μOR) Agonists
Corresponding authors: Bohua Zhong and Weiguo Shi (State Key Laboratory of Toxicology and Medical Countermeasures, Beijing Institute of Pharmacology & Toxicology, Beijing, China)
Molecules 2019, Volume 24, Issue 2 Page 259
Published online January 11th, 2019
https://doi.org/10.3390/molecules24020259
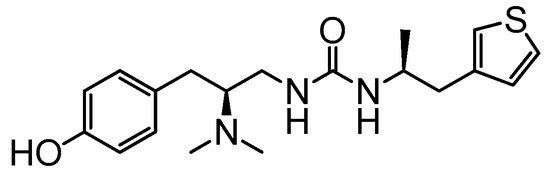
7d:

Corresponding authors: Bohua Zhong and Weiguo Shi (State Key Laboratory of Toxicology and Medical Countermeasures, Beijing Institute of Pharmacology & Toxicology, Beijing, China)
Molecules 2019, Volume 24, Issue 2 Page 259
Published online January 11th, 2019
https://doi.org/10.3390/molecules24020259
PZM21:‘Biased’ ligands of G protein-coupled receptors (GPCRs) represent a type of promising analgesic with reduced on-target side effects. PZM21, a potent μ-opioid-receptor (μOR)-biased agonist with a new chemical scaffold compared to classic opioids, has been identified as a therapeutic lead molecule for treating pain. In the current study, novel PZM21 analogues were synthesized and evaluated for their in vitro and in vivo efficacy. Novel compound 7a and PZM21 demonstrated undetectable β-arrestin-2 recruitment, however, their analgesic effects need to be further confirmed. Compounds 7b, 7d, and 7g were stronger analgesics than PZM21 in both the mouse formalin injection assay and the writhing test. Compound 7d was the most potent analogue, requiring a dose that was 1/16th to 1/4th of that of PZM21 for its analgesic activity in the two assays, respectively. Therefore, compound 7d could serve as a lead to develop new biased μOR agonists for treating pain.
7d:
Last edited:
S.J.B.
Bluelight Crew
- Joined
- Jan 22, 2011
- Messages
- 6,886
Synthesis and Structure–Affinity Relationships of Spirocyclic Benzopyrans with Exocyclic Amino Moiety
Corresponding author: Bernhard Wünsch (Institut für Pharmazeutische und Medizinische Chemie, Universität Münster, Münster, Germany)
Journal of Medicinal Chemistry 2019, Volume 62, Issue 8, Pages 4204–4217
Published online April 2nd, 2019
https://doi.org/10.1021/acs.jmedchem.9b00449

Corresponding author: Bernhard Wünsch (Institut für Pharmazeutische und Medizinische Chemie, Universität Münster, Münster, Germany)
Journal of Medicinal Chemistry 2019, Volume 62, Issue 8, Pages 4204–4217
Published online April 2nd, 2019
https://doi.org/10.1021/acs.jmedchem.9b00449
σ1 and/or σ2 receptors play a crucial role in pathological conditions such as pain, neurodegenerative disorders, and cancer. A set of spirocyclic cyclohexanes with diverse O-heterocycles and amino moieties (general structure III) was prepared and pharmacologically evaluated. In structure–activity relationships studies, the σ1 receptor affinity and σ1:σ2 selectivity were correlated with the stereochemistry, the kind and substitution pattern of the O-heterocycle, and the substituents at the exocyclic amino moiety. cis-configured 2-benzopyran cis-11b bearing a methoxy group and a tertiary cyclohexylmethylamino moiety showed the highest σ1 affinity (Ki = 1.9 nM) of this series of compounds. In a Ca2+ influx assay, cis-11b behaved as a σ1 antagonist. cis-11b reveals high selectivity over σ2 and opioid receptors. The interactions of the novel σ1ligands were analyzed on the molecular level using the recently reported X-ray crystal structure of the σ1 receptor protein. The protonated amino moiety forms a persistent salt bridge with E172. The spiro[benzopyran-1,1′-cyclohexane] scaffold and the cyclohexylmethyl moiety occupy two hydrophobic pockets. Exchange of the N-cyclohexylmethyl moiety by a benzyl group led unexpectedly to potent and selective μ-opioid receptor ligands.
Last edited:
S.J.B.
Bluelight Crew
- Joined
- Jan 22, 2011
- Messages
- 6,886
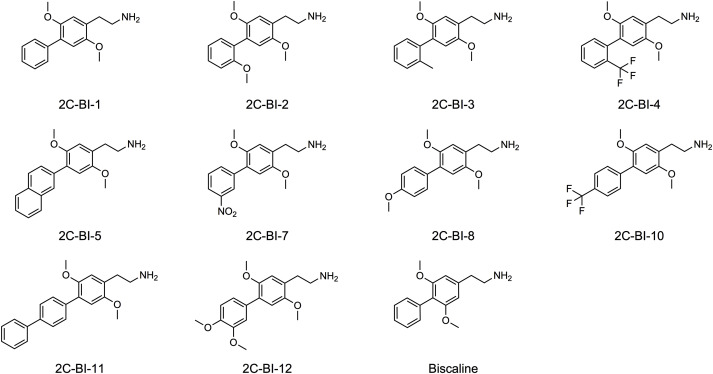
Monoamine receptor interaction profiles of 4-aryl-substituted 2,5-dimethoxyphenethylamines (2C-BI derivatives)
Corresponding author: Matthias E. Liechti (Division of Clinical Pharmacology and Toxicology, Department of Biomedicine, University of Basel, Basel, Switzerland)
European Journal of Pharmacology 2019, Volume 855, Pages 103-111
Published online May 4th, 2019
https://doi.org/10.1016/j.ejphar.2019.05.014
Corresponding author: Matthias E. Liechti (Division of Clinical Pharmacology and Toxicology, Department of Biomedicine, University of Basel, Basel, Switzerland)
European Journal of Pharmacology 2019, Volume 855, Pages 103-111
Published online May 4th, 2019
https://doi.org/10.1016/j.ejphar.2019.05.014
Many ring-substituted phenethylamines exert psychedelic effects that are thought to be primarily mediated by interactions with serotonergic 5-hydroxytryptamine 2 (5-HT2A) receptors. The 2,5-dimethoxyphenethylamine (2C derivative) core structure with small lipophilic substituents at the 4-position seems to be particularly favorable for psychedelic effects. In contrast, 2C derivatives with bulky lipophilic substituents at the 4-position of the phenyl ring tend to display antagonist behavior at serotonin 5-HT2 receptor sites. To gain a better understanding of agonist and antagonist behavior of substituted phenethylamines, binding affinities and functional activation and inhibition of a series of 4′-aryl substituted 2,5-dimethoxyphenethylamine (2C-BI derivatives) at various monoamine receptors were determined. In addition, the interactions of the compounds with monoamine transporters were assessed. Various 2C-BI derivatives potently bound to human serotonergic and adrenergic receptors and to rat and mouse trace amine-associated receptor 1. Additionally, 2C-BI-8 and 2C-BI-12 activated serotonin 5-HT2A and 5-HT2B receptors at submicromolar concentrations. 2C-BI-1 and 2C-BI-7 were the only 2C-BI derivatives to activate human trace amine-associated receptor 1. 2C-BI-3 and 2C-BI-4 interacted with monoamine transporters but with low overall potency. In conclusion, the tested 2C-BI derivatives displayed diverse pharmacological profiles. The relatively high affinities of various 2C-BI derivatives at the serotonin 5-HT2A receptor indicate a high steric tolerance of the binding pocket. Potent partial activation of the serotonin 5-HT2A receptor by 2C-BI-8 and 2C-BI-12 suggests that these substances may potentially exert psychedelic effects similar to other compounds of the 2C family.
sekio
Bluelight Crew
- Joined
- Sep 14, 2009
- Messages
- 21,994
7-Hydroxymitragynine Is an Active Metabolite of Mitragynine and a Key Mediator of Its Analgesic Effects
Andrew C. Kruegel, Rajendra Uprety, Steven G. Grinnell, Cory Langreck, Elizabeth A. Pekarskaya, Valerie Le Rouzic, Michael Ansonoff, Madalee M. Gassaway, John E. Pintar, Gavril W. Pasternak, Jonathan A. Javitch, Susruta Majumdar, Dalibor Sames
very cool
i wonder if cyp3a4 inducers would make kratom more effective...
Andrew C. Kruegel, Rajendra Uprety, Steven G. Grinnell, Cory Langreck, Elizabeth A. Pekarskaya, Valerie Le Rouzic, Michael Ansonoff, Madalee M. Gassaway, John E. Pintar, Gavril W. Pasternak, Jonathan A. Javitch, Susruta Majumdar, Dalibor Sames
... Here we report evidence that an active metabolite plays an important role in mediating the analgesic effects of mitragynine. We find that mitragynine is converted in vitro in both mouse and human liver preparations to the much more potent mu-opioid receptor agonist 7-hydroxymitragynine and that this conversion is mediated by cytochrome P450 3A isoforms. Further, we show that 7-hydroxymitragynine is formed from mitragynine in mice and that brain concentrations of this metabolite are sufficient to explain most or all of the opioid-receptor-mediated analgesic activity of mitragynine. At the same time, mitragynine is found in the brains of mice at very high concentrations relative to its opioid receptor binding affinity, suggesting that it does not directly activate opioid receptors. The results presented here provide a metabolism-dependent mechanism for the analgesic effects of mitragynine and clarify the importance of route of administration for determining the activity of this compound.
very cool
i wonder if cyp3a4 inducers would make kratom more effective...
Last edited: