Synthesis and Pharmacological Evaluation of Novel C‑8 Substituted Tetrahydroquinolines as Balanced-Affinity Mu/Delta Opioid Ligands for the Treatment of Pain
Corresponding author: Henry I. Mosberg (Department of Medicinal Chemistry, College of Pharmacy, University of Michigan, Michigan, United States)
ACS Chemical Neuroscience 2018, Volume 9, Issue 7, Pages 1840-1848
Published online April 20th, 2018
https://doi.org/10.1021/acschemneuro.8b00139
Abstract:
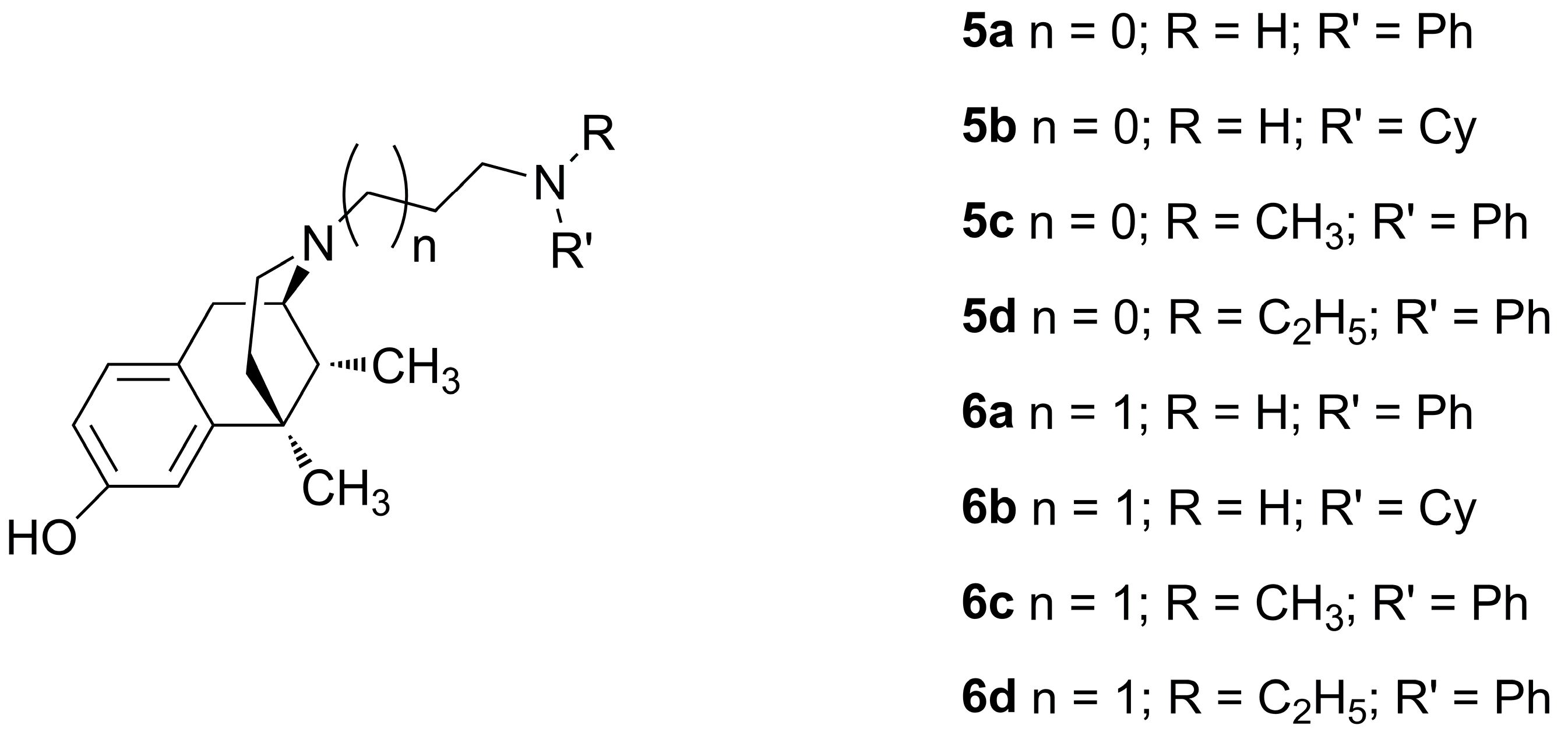
The use of opioids for the treatment of pain, while largely effective, is limited by detrimental side effects including analgesic tolerance, physical dependence, and euphoria, which may lead to opioid abuse. Studies have shown that compounds with a mu-opioid receptor (MOR) agonist/delta-opioid receptor (DOR) antagonist profile reduce or eliminate some of these side effects including the development of tolerance and dependence. Herein we report the synthesis and pharmacological evaluation of a series of tetrahydroquinoline-based peptidomimetics with substitutions at the C-8 position. Relative to our lead peptidomimetic with no C-8 substitution, this series affords an increase in DOR affinity and provides greater balance in MOR and DOR binding affinities. Moreover, compounds with carbonyl moieties at C-8 display the desired MOR agonist/DOR antagonist profile whereas alkyl substitutions elicit modest DOR agonism. Several compounds in this series produce a robust antinociceptive effect in vivo and show antinociceptive activity for greater than 2 h after intraperitoneal administration in mice.
Table 2. Effects of Alkyl and Halogen Substitutions on Affinity, Potency, and Efficacy
Table 3. Effects of Aryl, Carbonyl, and Amino Substitutions on Affinity, Potency, and Efficacy
Most of the compounds were tested for antinociceptive activity
in vivo via the mouse warm water tail withdrawal test (10 mg/kg, intraperitoneal injection). Compounds
7b,
c,
e, and
n were found to be fully efficacious,
7f was partially active, and the others has no activity.
---
It is worth noting that there is an FDA-approved drug for irritable bowel syndrome,
eluxadoline, which combines MOR agonism with DOR antagonism:
Has anyone had the chance to encounter this compound? If so, how does it compare to other opioids?