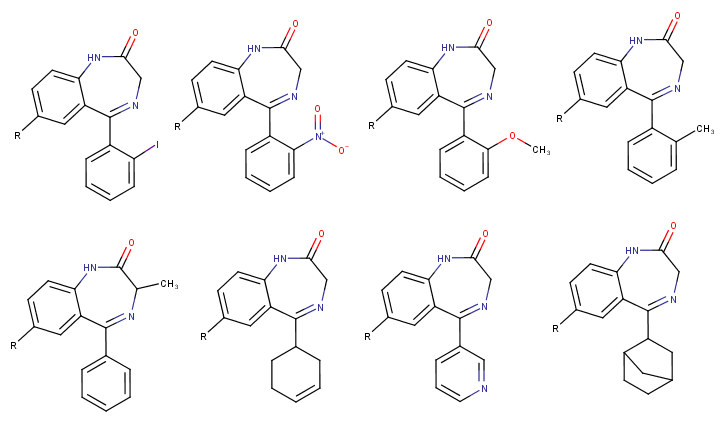
Does anyone know why certain theoretical possible benzodiazepine-analogues have never been made or generally considered? For example every halogen besides iodine is used, the nitro-unit is always on the phenyl-ring at the diazepine core and not on the other phenyl-unit (e.g. in benzos like flunitrazepam, clonazepam, nitrazepam etc.).
3-methyl benzos don't seem to exist besides meclonazepam (which is very good in my opinion, not as good as etizolam which is my nr.1 but it has similar qualities such as a "bright" effect (in contrast to "dark" ones like nifoxipam, lorazepam, phenazepam or all the RC -zolams with 2 halogens like Flubromazolam, Flualprazolam etc.), a decent half life which lingers minimally into the next day but without the known benzo depressions and it has a broad dosage-spectrum and recreational window as you can feel it from 1mg+ with light anxiolysis/disinhibition and the feeling of being "free" but even 10mg wont make you black out, just very wobbly as the muscle relaxation increases. I loved this effect also from etizolam, you feel heavily relaxed and comfortable but without this drowsy hypnotic sedation of some other benzos)
What about methoxy- or methyl substitutions on one of the phenyl rings? Would they still be active or generally too weak or something else? Could you replace the phenyl completely with something else like norbornane in the image below?
Or for example that cyclohexene-ring that is known from tetrazepam and the pyridine ring of bromazepam are only used in those two benzos except pyrazolam.
Btw., are there any benzos (pharmaceutical or from research papers etc.) that don't have any halogen substitutions at all?
If anyone knows something about this or has links to research papers or something similar I'd be thankful
")
(The "R" below on the molecules is just a placeholder for whatever substitution one could (also) make there)
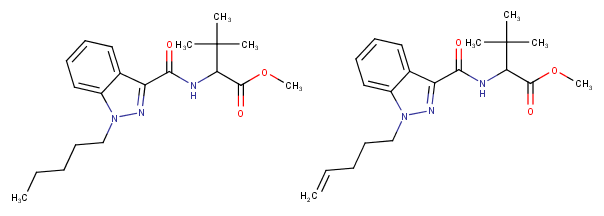
edit: I have another question; there are new cannabinoids on the market which have instead of the usual pentyl-side chain a "pentenyl"-chain. Can anyone guess what difference this creates? It looks like a too simple modification to be also safe as it hasn't been used until now, if it would be that easy than you could have just replaced the pentyl chains of the old cannabinoids with pentenyl's so I have the feeling that there must be a hitch to this kind of analogue, although it seems there are cannabinoids in the JWH-series that also have this kind of side chain but why is it only used now? Does it impact the molecule in a unforseeable way or make it more/less active?