-
N&PD Moderators: Skorpio | thegreenhand
-
Neuroscience & Pharmacology Discussion Welcome Guest
Posting Rules Bluelight Rules Recent Journal Articles Chemistry Mega-Thread FREE Chemistry Databases! Self-Education Guide
You are using an out of date browser. It may not display this or other websites correctly.
You should upgrade or use an alternative browser.
You should upgrade or use an alternative browser.
Are there any good SNDRIs yet?
- Thread starter atara
- Start date
checktest
Bluelighter
- Joined
- Jun 9, 2013
- Messages
- 338
I haven't been following too closely, but from some references it has been a bit bumpy.
It seems like a few times every year you will get papers (at various stages) like:

 www.ncbi.nlm.nih.gov
SMe1EC2M3 - Kinda tricyclic or carboline (?carbidine-esque, would be concerned about some channels) (Molecules)
www.ncbi.nlm.nih.gov
SMe1EC2M3 - Kinda tricyclic or carboline (?carbidine-esque, would be concerned about some channels) (Molecules)
www.ncbi.nlm.nih.gov
D-578 - Crossover PTSD.
A lot of the time you'll see developments from a group along structural motifs that generate some papers.
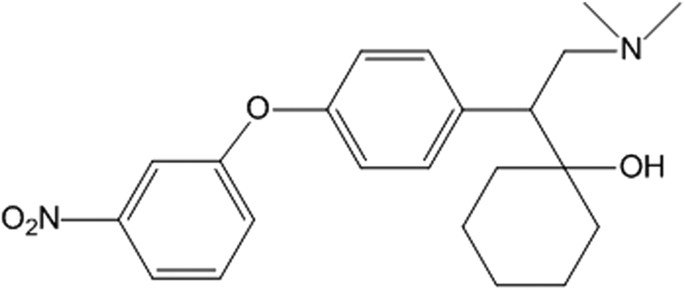
 www.nature.com
LPM580153 - Triple reuptake, (lowers body weight)
www.nature.com
LPM580153 - Triple reuptake, (lowers body weight)
www.ncbi.nlm.nih.gov
LPM580098 - Crossover neuropathic pain [Yantai]
www.ncbi.nlm.nih.gov
LPM570065 - SNDRI, increases testosterone in rats by ~37% [Yantai as above / Luye Pharma]
Some fairly recent available reviews:

 www.sciencedirect.com
Kinda has weird takes on some things
www.sciencedirect.com
Kinda has weird takes on some things
Ansofaxine I believe is undergoing trials right now, we'll see where it heads up
But the bodyweight effects mentioned highlight some tolerability problems (? toxicities) in trials that have popped up for SNDRIs in MDD (as well as new drug indications). Every one seems to be going for crossover or rare to gain some edge for clearance relative to competing against established antidepressants.
--I think the sertraline-related/development/metabolite dasotraline might get evaluated for binge eating. Still wonder if that gets approved whether they'll bring back tametraline or the other one.
--Tesofensine for obesity, probably binge eating.
--Amitfadine, which was looking promising circa 2013 kinda flopped in trials for MDD but still under for smoking/drinking.
--The development precursor to vortioxetine, tedatioxetine, I believe got stopped.
Not to mention emphasis on glutamatergic drugs and extensions for ketamine / combos rather than SNDRIs.
(The disappointing results of zuranolone put a damper on a couple of neurosteroids.)
 blogs.sciencemag.org
blogs.sciencemag.org
As fair as clinical, though, kinda some goofy triple stuff, at least where my psychiatrist is and some local practices.
Besides ketamine-related, SNRI-adjunct low-dose newer AAPs with DA partial agonism have been rising. (E.g. cariprazine, brexpiprazole). Taking the long way to basically get a less muscarinic tricyclic sometimes
Also I think I saw a trial of lower-dose long-acting stimulants on these AAPs as well (maybe Ritalin LA), which seems...interesting...
TLDR Meh
(TLDSNDRI? - Too Long to Develop SNDRIs?)
It seems like a few times every year you will get papers (at various stages) like:
Electrophysiology and Behavioral Assessment of the New Molecule SMe1EC2M3 as a Representative of the Future Class of Triple Reuptake Inhibitors - PubMed
SMe1EC2M3 is a pyridoindole derivative related to the neuroleptic drug carbidine. Based on the structural similarities of SMe1EC2M3 and known serotonin (5-HT), norepinephrine, and dopamine reuptake inhibitors, we hypothesized that this compound may also have triple reuptake inhibition efficacy...
www.ncbi.nlm.nih.gov
D-578, an orally active triple monoamine reuptake inhibitor, displays antidepressant and anti-PTSD like effects in rats - PubMed
Significant unmet needs exist for development of better pharmacotherapeutic agents for major depressive disorder (MDD) and post-traumatic stress disorder (PTSD) as the current drugs are inadequate. Our goal in this study is to investigate behavioral pharmacological characterization of a novel...
www.ncbi.nlm.nih.gov
A lot of the time you'll see developments from a group along structural motifs that generate some papers.
Antidepressant-like Effects of LPM580153, A Novel Potent Triple Reuptake Inhibitor - Scientific Reports
The purpose of this study was to characterize a novel compound, 4-[2-(dimethylamino)-1-(1-hydroxycyclohexyl) ethyl] phenyl 3-nitrophenyl ether, designated LPM580153. We used several well-validated animal models of depression to assess the antidepressant-like activity of LPM580153, followed by a...
www.nature.com
LPM580098, a Novel Triple Reuptake Inhibitor of Serotonin, Noradrenaline, and Dopamine, Attenuates Neuropathic Pain - PubMed
<span><b>Background and Purpose:</b> Sedation and somnolence remain serious adverse effects of the existing analgesics (e.g., pregabalin, duloxetine) for neuropathic pain. The available evidence indicates that serotonin (5-HT), noradrenaline (NE), and dopamine (DA) play important roles in...
www.ncbi.nlm.nih.gov
Acute, subchronic oral toxicity, and genotoxicity evaluations of LPM570065, a new potent triple reuptake inhibitor - PubMed
In the current study, to support the safety of LPM570065 as a new potent triple reuptake inhibitors (TRIs), LPM570065 was investigated through a single- and 13-week repeated-dose oral toxicity evaluation and mutagenicity assays. In an acute toxicity evaluation, Sprague-Dawley (SD) rats were...
www.ncbi.nlm.nih.gov
Some fairly recent available reviews:
The emergence of new antidepressants for clinical use: Agomelatine paradox versus other novel agents
This study was designed with the rational aim of discussing the emerging antidepressant agents that are likely to bring positive landmark, tremendous …
Ansofaxine I believe is undergoing trials right now, we'll see where it heads up
But the bodyweight effects mentioned highlight some tolerability problems (? toxicities) in trials that have popped up for SNDRIs in MDD (as well as new drug indications). Every one seems to be going for crossover or rare to gain some edge for clearance relative to competing against established antidepressants.
--I think the sertraline-related/development/metabolite dasotraline might get evaluated for binge eating. Still wonder if that gets approved whether they'll bring back tametraline or the other one.
--Tesofensine for obesity, probably binge eating.
--Amitfadine, which was looking promising circa 2013 kinda flopped in trials for MDD but still under for smoking/drinking.
--The development precursor to vortioxetine, tedatioxetine, I believe got stopped.
Not to mention emphasis on glutamatergic drugs and extensions for ketamine / combos rather than SNDRIs.
(The disappointing results of zuranolone put a damper on a couple of neurosteroids.)
Science | AAAS
As fair as clinical, though, kinda some goofy triple stuff, at least where my psychiatrist is and some local practices.
Besides ketamine-related, SNRI-adjunct low-dose newer AAPs with DA partial agonism have been rising. (E.g. cariprazine, brexpiprazole). Taking the long way to basically get a less muscarinic tricyclic sometimes
Also I think I saw a trial of lower-dose long-acting stimulants on these AAPs as well (maybe Ritalin LA), which seems...interesting...
TLDR Meh
(TLDSNDRI? - Too Long to Develop SNDRIs?)
Last edited:
- Joined
- Jul 2, 2008
- Messages
- 9,131
Not really. Effexor and Cymbalta at high doses may have some dopaminergic activity. But they're working on a DXM-bupropion combination medication, the former an SNRI and the latter an NDRI (but dopaminergic activity lower than that of stimulants).
They have some SNDRIs in trials.
Actually that's right: Zoloft has an SNDRI as a metabolite that may have some dopaminergic activity. More so at higher doses, as would follow.
They're wary of addiction and cardiac toxicity.
They have some SNDRIs in trials.
Actually that's right: Zoloft has an SNDRI as a metabolite that may have some dopaminergic activity. More so at higher doses, as would follow.
They're wary of addiction and cardiac toxicity.
SlowandFastandSlow
Bluelighter
- Joined
- Jan 21, 2020
- Messages
- 77
HDMP-28 and HDEP-28 were knocking around a few years back
polymath
Bluelight Crew
- Joined
- Nov 4, 2010
- Messages
- 1,884
I’ve been away from the field for a while, would anyone happen to have been following developments in this area?
(The obvious answer does not count)
What's the obvious answer? Bupropion? St John's wort? Or maybe the experimental drug that is mentioned at the end of SNDRI Wikipedia article.
Some compounds in the herb "skullcap" (Scutellaria baicalensis) are dopamine reuptake inhibitors without being positive reinforcing, and seem to protect against MPTP neurotoxicity and have an anti-ADHD effect in some animal model. Probably no one has tried combining those with conventional antidepressants. The lobeline alkaloids are another possibility.
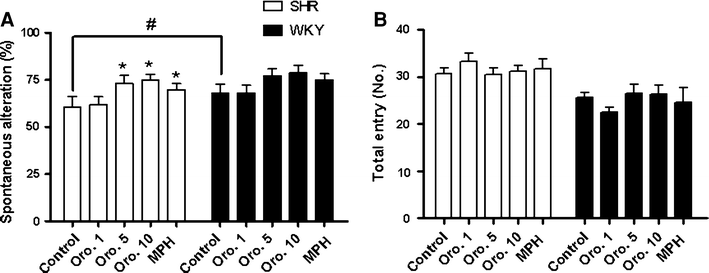
Oroxylin A improves attention deficit hyperactivity disorder-like behaviors in the spontaneously hypertensive rat and inhibits reuptake of dopamine in vitro - Archives of Pharmacal Research
In previous studies we have demonstrated that the γ-aminobutryic acid-A (GABA-A) receptor antagonist oroxylin A has an awakening effect and it also represses ADHD-like behaviors (hyperactivity, impulsivity and inattention) in the spontaneously hypertensive rat (SHR) model of attention-deficit...
 link.springer.com
link.springer.com
Edit: also, as the MAO-B inhibitor seleginine prevents MPTP-induced neural damage and has a lifespan-extending effect in animal experiments, maybe those non-addictive dopamine reuptake inhibitors could have this same benefit. Or maybe it's just the triple bond in selegiline being so good antioxidant/radical scavenger.
Last edited:
sekio
Bluelight Crew
- Joined
- Sep 14, 2009
- Messages
- 21,994
What's the obvious answer?
cocaine
The reason that MAO-B inhibitors prevent MPTP induced toxicity is very simple, because MPTP is not the active toxic species, MPP+ is, MPP+ is produced by monoamine oxidase action on MPTP. MPP+ is permanently charged and mitochondria seeking which is very redox active and so spins off all sorts of ROS whilst short circuiting mitochondrial energy generation. Some carbolines have a similar potency to generate redox active metabolites.
so blocking MAOB stops MPP+ formation. Blocking DAT stops MPP+ being taken up into the neurons so DAT inhibitors have the same protective action Amfonelic acid a highly selective and highly potent DAT inhibitor also prevents MPTP neurotoxicity at very low doses, something which has been investigated as an anti parkinson drug. However MPTP induced lesions are a very poor model for real parkinsons disease.
MPTP also damges NE neurons but the evidence is not so clear cut whether NET is transporting MPP+ or whether the fairly lipophillic MPTP can passively diffuse in sufficient concentrations to be toxic in NET expressing neurons.
The reason seleginine has lifespan extending effects in rats, and also insects subjected to stressing factors is currently being investigated at several locations. It has a pronounced tumor supressing action in pure bred, mostly homozygous, rats which a major contributor to extended longevity. But the action in the stress induced mortality model in insects seems different. It also does not appear to have much to be to do with MAOB inhibition either. It is not to do with simple antioxidant action, N-Propargyl is a relatively poor antioxidant it is surprisingly stable in vivo but is cleaved eventually however it is a much better suicide enzyme inhibitor for a variety of enzymes. That is all I can say at this point
so blocking MAOB stops MPP+ formation. Blocking DAT stops MPP+ being taken up into the neurons so DAT inhibitors have the same protective action Amfonelic acid a highly selective and highly potent DAT inhibitor also prevents MPTP neurotoxicity at very low doses, something which has been investigated as an anti parkinson drug. However MPTP induced lesions are a very poor model for real parkinsons disease.
MPTP also damges NE neurons but the evidence is not so clear cut whether NET is transporting MPP+ or whether the fairly lipophillic MPTP can passively diffuse in sufficient concentrations to be toxic in NET expressing neurons.
The reason seleginine has lifespan extending effects in rats, and also insects subjected to stressing factors is currently being investigated at several locations. It has a pronounced tumor supressing action in pure bred, mostly homozygous, rats which a major contributor to extended longevity. But the action in the stress induced mortality model in insects seems different. It also does not appear to have much to be to do with MAOB inhibition either. It is not to do with simple antioxidant action, N-Propargyl is a relatively poor antioxidant it is surprisingly stable in vivo but is cleaved eventually however it is a much better suicide enzyme inhibitor for a variety of enzymes. That is all I can say at this point
Last edited:
Nicomorphinist
Bluelighter
- Joined
- Apr 18, 2019
- Messages
- 1,401
The ethylenediamine antihistamine tripelennamine, so beloved of opioid users myself included for its potentiating actions, is a weak to middling and dirty (not so selective) treble reuptake inhibitor (serotonin, norepinephrine, dopamine) and was the basis for the research which yielded fluoxetine and illuminated the first-generation antihistamine to modern reuptake type antidepressant route . . . since it already has these effects and only about 7 per cent of the anti-muscarinic strength of atropine, how about tripelennamine itself or a close analogue perhaps mixed with something to combat the sedation, which is not that bad, actually. The anti-muscarinic effect may be part of it -- I get even better effects with adding orphenadrine to tripelennamine by itself and my morphine-tripelennamine mixture and orphenadrine and other first-generation antihistamines actually list euphoria as a side effect in their descriptions in reference works and monographs. Tripelennamine included, of course.
checktest
Bluelighter
- Joined
- Jun 9, 2013
- Messages
- 338
But they're working on a DXM-bupropion combination medication
Yeah, that had a few trials in Dec. Here's a post
AXS-05 did end up having a successful phase III trial, though I haven't read the full details. M Fava blurb.
Press junk
Axsome Therapeutics Announces AXS-05 Achieves Primary Endpoint in GEMINI Phase 3 Trial in Major Depressive Disorder | BioSpace
And of course they have esbupropion-DXM in the pipeline. Gotta get that juicy isomer extension.
ClinicalTrials.gov
 clinicaltrials.gov
clinicaltrials.gov
I mean if we're talking nonselective or bad SNDRIs nefazodone can still be a possibility, haha. And massive doses of venlafaxine. Not to mention methylphenidate-SSRI/SNRI and such combos.
I've been wondering with the solriamfetol approval whether they'll start on a couple less selective NDRIs and methylphenidate analogs like the HDMP. Not going to sign up for naphthalene though.
Gotta pass abuse trials.
Last edited:
polymath
Bluelight Crew
- Joined
- Nov 4, 2010
- Messages
- 1,884
The ethylenediamine antihistamine tripelennamine, so beloved of opioid users myself included for its potentiating actions, is a weak to middling and dirty (not so selective) treble reuptake inhibitor (serotonin, norepinephrine, dopamine) and was the basis for the research which yielded fluoxetine and illuminated the first-generation antihistamine to modern reuptake type antidepressant route . . . since it already has these effects and only about 7 per cent of the anti-muscarinic strength of atropine, how about tripelennamine itself or a close analogue perhaps mixed with something to combat the sedation, which is not that bad, actually. The anti-muscarinic effect may be part of it -- I get even better effects with adding orphenadrine to tripelennamine by itself and my morphine-tripelennamine mixture and orphenadrine and other first-generation antihistamines actually list euphoria as a side effect in their descriptions in reference works and monographs. Tripelennamine included, of course.
Some old pharmacopoeia book I used to read said that orphenadrine also has some kind of psychostimulant effect that increases its anti-parkinsonian effectiveness... I haven't found any other source claiming it to inhibit dopamine reuptake, maybe the author of that book confused it with tripelennamine. Some rare people have been known to abuse anticholinergics like biperiden and tricyclics because they have a mood-lifting effect in small doses (no one probably enjoys the delirium from larger amounts). I think the reason why tripelennamine was mixed with pentazocine by some opiate users was that the antihistaminic/anticholinergic only potentiates the mu receptor effect but not the dysphoric kappa one.
The late 1990s "DXM FAQ" claimed that DXM also inhibits DA reuptake, similar to bupropion, but no present-day source seems to say the same.
Nicomorphinist
Bluelighter
- Joined
- Apr 18, 2019
- Messages
- 1,401
Some old pharmacopoeia book I used to read said that orphenadrine also has some kind of psychostimulant effect that increases its anti-parkinsonian effectiveness... I haven't found any other source claiming it to inhibit dopamine reuptake, maybe the author of that book confused it with tripelennamine. Some rare people have been known to abuse anticholinergics like biperiden and tricyclics because they have a mood-lifting effect in small doses (no one probably enjoys the delirium from larger amounts). I think the reason why tripelennamine was mixed with pentazocine by some opiate users was that the antihistaminic/anticholinergic only potentiates the mu receptor effect but not the dysphoric kappa one.
The late 1990s "DXM FAQ" claimed that DXM also inhibits DA reuptake, similar to bupropion, but no present-day source seems to say the same.
Orphenadrine and I believe also tripelennamine affect the NMDA receptor and nociception system too and that added to the antihistamine and anticholinergic action are what make them both so good with opioids, and the overall euphoric effect of tripelennamine could definitely overcome the dysphoria from the pentazocine's κ opioid effects. All the more adding orphenadrine as a third ingredient. This happened even more so with phenazocine, four times as strong as pentazocine, and dezocine, maybe 50 per cent stronger. The tripelennamine and phenazocine was the most euphoric and analgesic of the three mixtures.
Given how dirty (in the pharmacodynamic sense) orphenadrine is especially and also tripelennamine, who knows if 20 years if there is an omicron (ο opioid) and tau (τ opioid) receptor discovered in them . . .
The other agonist-antagonist and/or μ opioid partial agonists have tried ths with include the phenazepanes -- I have tried tripelennamine and tripelennamine plus orphenadrine with meptazinol and ethoheptazine, but not with proheptazine, and the effect with meptazinol in particular was that it felt more euphoric, and the same with the benzenoids tramadol and tapentadol.
Since orphenadrine lasts for 8 hours, I have tripelennamine and orphenadrine mixed with every other hydromorphone shot each day and the lift is right about like morphine, with which I mixed the two special ingredients, and it also works well with doing it with nicomorphine and dihydromorphine. Blue Velvet is the grandfather of Ts & Blues, invented in the early 1950s, it is tripelennamine plus morphine. In the 1980s, people started mixing pentazocine neat with tripelennamine and methylphenidate and it was called Kibbles & Bits . . . The hydromorphone/morphine/nicomorphine and tripelennamine and orphenadrine ménage à trois makes my mouth suddenly go dry, which I actually like because it means that about 10 seconds away is the beginning of a very rapid increase in the euphoria (bang) and physical effects (rush) including a muted itchy effect and the rush somehow modified for the better or a wash.
People also take conservative doses of hyoscine (scopolamine) for euphoria, and for breakthrough pain I get the old recipe of Scophedal compounded: Scopolamine + ephedrine + eukodal (injectable oxycodone) -- the wonder drug of the 1930s marketed by Merck as SEE (Scopolamine, Ephedrine, Eukodal) to 1940 and as Scophedal from 1940 to 1987. Dr Kirschner, the inventor, and the manufacturers wrote in the monograph that it produces "very deep analgesia and profound euphoria" which I can avouch and was used on the battlefield in the II. World War for this reason as well as the kind of uses propofol and morphine + midazolam are put today. The idea was to keep vital signs as close to normal as possible, which I can also report.
Another interesting antihistamine is phenindamine, a first-generation piperidine antihistamine which was marketed as Theophorin and was the shot that William S Burroughs got at the Lexington Narcotics Farm which he said "felt like M" this of course whilst withdrawal was going on . . .and indeed there has been research about using orphenadrine to help with opioid withdrawal.
Some of the original advertising in the 1950s also said that orphenadrine HCl had a useful anti-depressant effect which helped it to work against muscle spams and pain:
www.decodog.com/inven/MD/md30554.jpg

Last edited:
polymath
Bluelight Crew
- Joined
- Nov 4, 2010
- Messages
- 1,884
^ Before modern antipsychotics were invented, a mixture called scopomorphine (scopolamine and morphine) was used for sedating psychiatric patients. It put people in a docile state and probably also made them less likely to piss in their pants when in restraints (anticholinergics cause urinary retention). The downside is that some patients could have intentionally behaved aggressively to get a shot of that drug.
Edit: In this article, it turns out that diphenhydramine and chlorpheniramine also have a dopamine reuptake inhibiting effect:
 onlinelibrary.wiley.com
onlinelibrary.wiley.com
Many of the sedative antihistamines have some structural similarity to benzylpiperazine. Maybe if you extract the antihistamines from pills and vape them like crack, they would even cause an euphoric "rush". Or at least something substantial enough for you to actually notice it.
Edit: In this article, it turns out that diphenhydramine and chlorpheniramine also have a dopamine reuptake inhibiting effect:
Error - Cookies Turned Off
Many of the sedative antihistamines have some structural similarity to benzylpiperazine. Maybe if you extract the antihistamines from pills and vape them like crack, they would even cause an euphoric "rush". Or at least something substantial enough for you to actually notice it.
Last edited:
ChemicallyEnhanced
Bluelighter
- Joined
- Apr 29, 2018
- Messages
- 9,495
Not really. Effexor and Cymbalta at high doses may have some dopaminergic activity. But they're working on a DXM-bupropion combination medication, the former an SNRI and the latter an NDRI (but dopaminergic activity lower than that of stimulants).
They have some SNDRIs in trials.
Actually that's right: Zoloft has an SNDRI as a metabolite that may have some dopaminergic activity. More so at higher doses, as would follow.
They're wary of addiction and cardiac toxicity.
Yeah, Zoloft does act as a DRI but only at doses of 300mg+
Bravoncius Roxford
Bluelighter
- Joined
- Feb 1, 2017
- Messages
- 105
I’ve been away from the field for a while, would anyone happen to have been following developments in this area?
(The obvious answer does not count)
The problem with SNDRIs is that it is extremely difficult to come up with molecules that have high affinities for all three transporters due to the differences in amino acid configurations of all three proteins. Even cocaine, the "prototypical" SNRDI inhibitor is not really a SNDRI, but rather a drug that has many different enantiomers, with some having more affinities for one transporter over the other 2. It is exceptionally easy to come up with drugs that inhibited both the dopamine and norepinephrine transporters, because of the molecular similarity between the two transporters. In fact, it is much harder to come up with selective norepinephrine and dopamine reuptake inhibitors than it is to come with non-selective DA and NE reuptake inhibitors. The two catecholamines are very similar, and so are the transporters, so drugs that inhibit one also inhibit the other to one degree or another. This is why bupropion, which was created to be a NRIs, is effective for treating the anedonic aspects of depression at high doses, at it also inhibits the reuptake of dopamine. Of course, bupropion is a very weak dopamine reuptake inhibitor(IC50 6 Um), and even at huge doses it only has mild DA reuptake inhibitor properties. But large doses of bupropion do increase extracellular DA levels to some extent. The prototypical "pure" NE and DA reuptake inhibitors are, respectively, reboxetine and amineptine. But both drugs also increase the reuptake of the other catecholamine at high doses: doses of 10+ milligrams of reboxetine cause significant DA reuptake inhibition, and doses of amineptine of 200+ milligrams cause significant NE reuptake inhibition.
The big problem when you try to come up with reuptake inhibitors for all 3 monoamines is the molecular dissimilarity between norepinephrine and dopamine, on the one hand, and serotonin on the other. NE and DA are catecholamines, while serotonin is an indolamine. This means that the transporters for the catecholamines and for the indolamine serotonin will be quite different proteins. You can still find rare molecules that can on occasion inhibit the reuptake of one of the catecholamines and serotonin at once. A good example would be sertraline, the antidepressant, which inhibits the reuptake of both serotonin and dopamine. Of course, because sertraline has such extremely high binding to albumin in blood, it does not inhibit the reuptake of DA at clinical doses, despite being as potent as methylphenidate for DA reuptake inhibition. But molecules with affinity for the serotonin and norepinephrine and dopamine transporters are quite difficult to come by.
There is another problem, also: for reasons that one does not understand, triple reuptake inhibitors have a tendency to cause serious serotonin syndrome. This should not be expected, as the DA and NE transporters have no affinity for ST, and inhibition of the ST transporter alone usually doesn't cause serotonin syndrome. Serotonin syndrome, which results when the elevations of extracellular serotonin are massive, usually requires the combination of inhibition of two mechanisms that strongly controls the amount of serotonin in the extracellular space. Most ST syndromes, especially the severe ones that can kill you, result when you inhibit the serotonin transporter while at the same time inhibiting the catabolism of serotonin by inhibiting MAO-A. That is, severe ST syndrome results from taking a strong SRI with a strong MAO-A inhibitor. Really strong SRI would be paroxetine, clomipramine and escitalopram, all with Ki at 1 nmol or lower. And potent MAO-A inhibitors would be phenelzine and isocarboxazid, all with IC50 for inhibiting MAO-A below 1 Ug. Moclobemide also produces serious ST syndrome when combined with a SRI, but because moclobemide is fairly weak and reversible, death is usually not expected. The really lethal ST result from combining paroxetine, clomipramine or escitalopram with the old non-selective MAOIs like phenelzine or tranylcypromine. Just taking one 20 mg pill or paroxetine or escitalopram while on 60 mg of phenelzine a day can kill you in a matter of hours. But you wouldn't expect ST syndrome from a triple reuptake inhibitor when taken alone, since MAO-A remains active for breaking down excess serotonin. But this is not what happens. Usually, triple reuptake inhibitors produce serious ST syndrome even when taken as a solo drug.
As far as I know, the only triple reuptake inhibitor that was designed for human use was liafensine. However, it failed to show superior effectiveness to SNRIs. It was slightly more effective than the SSRIs, but not better than the SNRIs like duloxetine. This, and slso the fact that liafensine induced serotonin syndrome with fever and dystonia at clinical doses, canned the drug for human use.
If you want to know what it's like to experience significant elevations of all the 3 major neurotransmitter at once, then you should just take the non-selective irreversible inhibitors or just take cocaine. But of course, not non-selective MAOIs with cocaine because that is suicidal. How does a stroke sound? I mean either one or the other. Or you could try escitalopram + reboxetine + amineptine. While there are no significant liver enzyme interactions here, combining 3 drugs at once would be quite hard on your system. Or you could just try escitalopram + 300 mg amineptine. The latter significantly inhibits NE reuptake at those doses, so you would experience elevations of all 3 neurotransmitters. But, again, the whole ST syndrome makes this quite dangerous.
checktest
Bluelighter
- Joined
- Jun 9, 2013
- Messages
- 338
I thought reboxetine and atomoxetine had more SERT activity relative to DAT. I mean still relatively selective for NET but yeah.
NET, DAT, and SERT are all part of the solute carrier 6A family (thus SLC6A 2,3,4) respectively, and have strong similarities. What binding sites for particular drugs (let alone questions of biased agonism or other downstream changes) and actual dynamics are interesting. How have our example ligands biased our own research.

 www.ncbi.nlm.nih.gov
www.ncbi.nlm.nih.gov
www.ncbi.nlm.nih.gov
Covering those sites at similarly affinities selectively without binding to anti-targets like 5-ht2b agonism , hERG and some adrenergic problems can be an issue. I mean nefazodone is pretty close to 1:1:1 but had those liver side effects and a dirty metabolite in mcpp, as well as adrenergic effects.
Also whether a drug is really a reuptake inhibitor vs some type of releaser or reverse mechanism is a component risk for serotonin syndrome. Pure reuptake inhibition less than release, though many interactions. (Say a 5-ht2a antagonism opposing some serotonergic effects...).
And DA type abuse.
(Forgot nortriptyline + sertraline as a combo. )
I mean psychs will do some pretty strong things. Stimulants added on to non-selective MAOIs used to be more common (but as such the stroke risk/hypertensive crises/5-ht syndrome possibilities due to individual and dietary factors.) Sometimes they did tricyclics before/with MAOIs for some side effect modulation.
[I was on 70 mg tranylcypromine and 10-20mg methylphenidate, and even low dose buspirone with that. I do remember looking at my old bottles of 80mg prozac and desipramine and thinking how that would go at the hospital...anyways]
Still, good SNDRIs though would be interesting
NET, DAT, and SERT are all part of the solute carrier 6A family (thus SLC6A 2,3,4) respectively, and have strong similarities. What binding sites for particular drugs (let alone questions of biased agonism or other downstream changes) and actual dynamics are interesting. How have our example ligands biased our own research.
The solute carrier 6 family of transporters
The solute carrier 6 (SLC6) family of the human genome comprises transporters for neurotransmitters, amino acids, osmolytes and energy metabolites. Members of this family play critical roles in neurotransmission, cellular and whole body homeostasis. Malfunction ...
www.ncbi.nlm.nih.gov
The SLC6 transporters: perspectives on structure, functions, regulation, and models for transporter dysfunction - PubMed
The human SLC6 family is composed of approximately 20 structurally related symporters (co-transporters) that use the transmembrane electrochemical gradient to actively import their substrates into cells. Approximately half of the substrates of these transporters are amino acids, with others...
www.ncbi.nlm.nih.gov
Covering those sites at similarly affinities selectively without binding to anti-targets like 5-ht2b agonism , hERG and some adrenergic problems can be an issue. I mean nefazodone is pretty close to 1:1:1 but had those liver side effects and a dirty metabolite in mcpp, as well as adrenergic effects.
Also whether a drug is really a reuptake inhibitor vs some type of releaser or reverse mechanism is a component risk for serotonin syndrome. Pure reuptake inhibition less than release, though many interactions. (Say a 5-ht2a antagonism opposing some serotonergic effects...).
And DA type abuse.
(Forgot nortriptyline + sertraline as a combo. )
I mean psychs will do some pretty strong things. Stimulants added on to non-selective MAOIs used to be more common (but as such the stroke risk/hypertensive crises/5-ht syndrome possibilities due to individual and dietary factors.) Sometimes they did tricyclics before/with MAOIs for some side effect modulation.
[I was on 70 mg tranylcypromine and 10-20mg methylphenidate, and even low dose buspirone with that. I do remember looking at my old bottles of 80mg prozac and desipramine and thinking how that would go at the hospital...anyways]
Still, good SNDRIs though would be interesting
Last edited:
atara
Bluelighter
- Joined
- Apr 1, 2010
- Messages
- 2,785
In my idleness I had forgotten that there are actually two different categories of answers to this question.
There is the question about recreational SNDRIs, which would likely have affinities D > N ~ S
And there is the question about antidepressant SNDRIs, which would almost certainly have S > D ~ N (and probably N > D to minimize abuse)
I admit, I was rather more concerned about the former question, but I'm impressed by the work done to attack the latter.
See? Gentle as a lamb.
As for the latter question, this is a much more difficult nut to crack of course because it is a more difficult problem to solve. We have lots of ways to get high, we have few ways to treat depression that does not respond to conventional antidepressants. Furthermore there is the tension between what is an abused chemical versus an effective antidepressant. If we require N > D so that you won't enjoy getting high, then we incur potential N-related toxicity at therapeutic levels of D. If not, the drug is probably abusable, or we have S >> D which leads to S-related toxicity. Checking the list of compounds on Wikipedia we do indeed see a lot of S > N > D and a few surprising cases of N > S > D and N > D > S. In practically all cases N > D.
Unfortunately though if you're looking at affinities where S, N = 3-10x D, you're not really fundamentally changing much from SNRI-land, so I wonder if you can ever get much of a change in who is treated effectively by this drug category than you did with the SNRIs.
The general assertion that SND-affinity is too hard seems suspect. MDMA and AET display this binding pattern, albeit as releasers. Plus while S and N are rather different and D much like N, the SNRI drug class has really taken off. I could imagine, though, that if you bind to receptors for all three of these, you might also bind easily to MAO.
From the viewpoint of a practitioner, when you have some depressed patients, about 2/3 of them will respond quickly to SSRIs. You're used to SSRIs and you know how to deal with their side effects, but you still wish you could treat the last third of patients. The pharmaceutical companies basically seem to be trying to make the SSRIs stronger: SNRIs treat some more patients but have more side effects. SNDRIs could be the next step according to this theory. But implicitly what we're saying is "the depression didn't respond to SSRIs because it's really bad so we need to make it stronger", where when I say "make it stronger" we have a drug that's still an SSRI but with some extra activity.
But we might question the idea that SSRI-resistant depression is specifically a stronger version of SSRI-responsive depression. We could easily be looking at two (or fifty-seven) underlying etiologies. In other words, the patients who don't respond to SSRIs might respond to completely different drug actions, rather than more of them, because mental disorders are weird, and depression, in particular, is similar enough to some normal functions of the mind that it might be induced by a variety of different things. (This applies also to anxiety, but not to, e.g., schizophrenia.)
The only truly weird antidepressants I can think of like that are rubidium, agomelatine, tianeptine, and SAMe. But SAMe's potential to contribute to serotonin syndrome suggests that it, too, acts like an SSRI without being one. Agomelatine is the most promising, because it's MOA is different and probably unrelated to its liver toxicity. Rubidium is too expensive. Tianeptine is abuse-prone.
But maybe the solution to treatment-resistant depression lies in finding more efficient ways to separate rubidium from potassium in salt mines. Who would have thought?
But phenyltropanes are hard to make ;_;
Mazindol et alii are new to me. I'm not aware of the history of this drug class. It's intriguing.
Now there's something I want to try!
There is the question about recreational SNDRIs, which would likely have affinities D > N ~ S
And there is the question about antidepressant SNDRIs, which would almost certainly have S > D ~ N (and probably N > D to minimize abuse)
I admit, I was rather more concerned about the former question, but I'm impressed by the work done to attack the latter.
Come to think of it, I think that the naproxen-shaped analog of HDMP-28 should have less hepatotoxicity than the parent compound. Naproxen holds a naphthalene ring with a 6-methoxy group and is metabolized exclusively at the 6-methoxy group. It has undergone rigorous safety testing and is OTC approved despite that foreboding moiety.I've been wondering with the solriamfetol approval whether they'll start on a couple less selective NDRIs and methylphenidate analogs like the HDMP. Not going to sign up for naphthalene though.
See? Gentle as a lamb.
As for the latter question, this is a much more difficult nut to crack of course because it is a more difficult problem to solve. We have lots of ways to get high, we have few ways to treat depression that does not respond to conventional antidepressants. Furthermore there is the tension between what is an abused chemical versus an effective antidepressant. If we require N > D so that you won't enjoy getting high, then we incur potential N-related toxicity at therapeutic levels of D. If not, the drug is probably abusable, or we have S >> D which leads to S-related toxicity. Checking the list of compounds on Wikipedia we do indeed see a lot of S > N > D and a few surprising cases of N > S > D and N > D > S. In practically all cases N > D.
Unfortunately though if you're looking at affinities where S, N = 3-10x D, you're not really fundamentally changing much from SNRI-land, so I wonder if you can ever get much of a change in who is treated effectively by this drug category than you did with the SNRIs.
The general assertion that SND-affinity is too hard seems suspect. MDMA and AET display this binding pattern, albeit as releasers. Plus while S and N are rather different and D much like N, the SNRI drug class has really taken off. I could imagine, though, that if you bind to receptors for all three of these, you might also bind easily to MAO.
From the viewpoint of a practitioner, when you have some depressed patients, about 2/3 of them will respond quickly to SSRIs. You're used to SSRIs and you know how to deal with their side effects, but you still wish you could treat the last third of patients. The pharmaceutical companies basically seem to be trying to make the SSRIs stronger: SNRIs treat some more patients but have more side effects. SNDRIs could be the next step according to this theory. But implicitly what we're saying is "the depression didn't respond to SSRIs because it's really bad so we need to make it stronger", where when I say "make it stronger" we have a drug that's still an SSRI but with some extra activity.
But we might question the idea that SSRI-resistant depression is specifically a stronger version of SSRI-responsive depression. We could easily be looking at two (or fifty-seven) underlying etiologies. In other words, the patients who don't respond to SSRIs might respond to completely different drug actions, rather than more of them, because mental disorders are weird, and depression, in particular, is similar enough to some normal functions of the mind that it might be induced by a variety of different things. (This applies also to anxiety, but not to, e.g., schizophrenia.)
The only truly weird antidepressants I can think of like that are rubidium, agomelatine, tianeptine, and SAMe. But SAMe's potential to contribute to serotonin syndrome suggests that it, too, acts like an SSRI without being one. Agomelatine is the most promising, because it's MOA is different and probably unrelated to its liver toxicity. Rubidium is too expensive. Tianeptine is abuse-prone.
But maybe the solution to treatment-resistant depression lies in finding more efficient ways to separate rubidium from potassium in salt mines. Who would have thought?
RTI-xxx have some pretty nice SNDRIs, homomazindol maybe too.
But phenyltropanes are hard to make ;_;
Mazindol et alii are new to me. I'm not aware of the history of this drug class. It's intriguing.
a mixture called scopomorphine (scopolamine and morphine) was used for sedating psychiatric patients.
Now there's something I want to try!
Nicomorphinist
Bluelighter
- Joined
- Apr 18, 2019
- Messages
- 1,401
In my idleness I had forgotten that there are actually two different categories of answers to this question.
There is the question about recreational SNDRIs, which would likely have affinities D > N ~ S
And there is the question about antidepressant SNDRIs, which would almost certainly have S > D ~ N (and probably N > D to minimize abuse)
I admit, I was rather more concerned about the former question, but I'm impressed by the work done to attack the latter.
Come to think of it, I think that the naproxen-shaped analog of HDMP-28 should have less hepatotoxicity than the parent compound. Naproxen holds a naphthalene ring with a 6-methoxy group and is metabolized exclusively at the 6-methoxy group. It has undergone rigorous safety testing and is OTC approved despite that foreboding moiety.
See? Gentle as a lamb.
As for the latter question, this is a much more difficult nut to crack of course because it is a more difficult problem to solve. We have lots of ways to get high, we have few ways to treat depression that does not respond to conventional antidepressants. Furthermore there is the tension between what is an abused chemical versus an effective antidepressant. If we require N > D so that you won't enjoy getting high, then we incur potential N-related toxicity at therapeutic levels of D. If not, the drug is probably abusable, or we have S >> D which leads to S-related toxicity. Checking the list of compounds on Wikipedia we do indeed see a lot of S > N > D and a few surprising cases of N > S > D and N > D > S. In practically all cases N > D.
Unfortunately though if you're looking at affinities where S, N = 3-10x D, you're not really fundamentally changing much from SNRI-land, so I wonder if you can ever get much of a change in who is treated effectively by this drug category than you did with the SNRIs.
The general assertion that SND-affinity is too hard seems suspect. MDMA and AET display this binding pattern, albeit as releasers. Plus while S and N are rather different and D much like N, the SNRI drug class has really taken off. I could imagine, though, that if you bind to receptors for all three of these, you might also bind easily to MAO.
From the viewpoint of a practitioner, when you have some depressed patients, about 2/3 of them will respond quickly to SSRIs. You're used to SSRIs and you know how to deal with their side effects, but you still wish you could treat the last third of patients. The pharmaceutical companies basically seem to be trying to make the SSRIs stronger: SNRIs treat some more patients but have more side effects. SNDRIs could be the next step according to this theory. But implicitly what we're saying is "the depression didn't respond to SSRIs because it's really bad so we need to make it stronger", where when I say "make it stronger" we have a drug that's still an SSRI but with some extra activity.
But we might question the idea that SSRI-resistant depression is specifically a stronger version of SSRI-responsive depression. We could easily be looking at two (or fifty-seven) underlying etiologies. In other words, the patients who don't respond to SSRIs might respond to completely different drug actions, rather than more of them, because mental disorders are weird, and depression, in particular, is similar enough to some normal functions of the mind that it might be induced by a variety of different things. (This applies also to anxiety, but not to, e.g., schizophrenia.)
The only truly weird antidepressants I can think of like that are rubidium, agomelatine, tianeptine, and SAMe. But SAMe's potential to contribute to serotonin syndrome suggests that it, too, acts like an SSRI without being one. Agomelatine is the most promising, because it's MOA is different and probably unrelated to its liver toxicity. Rubidium is too expensive. Tianeptine is abuse-prone.
But maybe the solution to treatment-resistant depression lies in finding more efficient ways to separate rubidium from potassium in salt mines. Who would have thought?
But phenyltropanes are hard to make ;_;
Mazindol et alii are new to me. I'm not aware of the history of this drug class. It's intriguing.
Now there's something I want to try!
Morphine and opium were the prototype anti-depressants until the 1950s and scopolamine was used for tranquillisation, well, it is even today, and the mixture is indeed groovy.
It was used in obstetric anaesthesia too until it was slowly replaced by pethidine, piminodine, anileridine, alphaprodine &c in the 1950s, and as described in this or another thread, scopolamine with oxycodone and ephedrine is a good mixture for all sorts of things. If I take it for severe breakthrough pain more than twice or three times a day especially mixed with tripelennamine, orphenadrine, and caffeine it gets hard to know what day it is. I have a bunch of people around, de facto trip sitters I guess one could say, yet the lost time feeling such as being in bad shape on a Wednesday morning and suddenly find myself in the late afternoon on Friday. That was my first thought in re the lines:
I've been sitting here since Wednesday morning
Wednesday morning can't believe my ears
"Jazz Police" verse 1
Leonard Cohen 1988
Last edited:
Nicomorphinist
Bluelighter
- Joined
- Apr 18, 2019
- Messages
- 1,401
Naproxen especially has psychoactive effects (listed as euphoria and emotional lability on package inserts) and I am wondering if there is a prevailing theory as to why this is. I can vouch for the effect. I always thought that being out of pain would make one happier, and it does, but there is a clear mood lift associated with naproxen which can proceed to giddiness and can help out narcotics by contributing to analgesia, but I doubt the mechanism is opioid like. I notice it with ketoprofen and ibuprofen too, but not piroxicam, diclofenac, but to some extent I do with ketorolac. When we consider all of the information above, including the analgesic effect of first generation antidepressants and others, I would think it was obvious that euphoria is part of analgesia and no drug truly separating them will ever be discoverered. In fact venlafaxine and clonidine have quasi-narcotic effects that prisoners especially use.
Would nefopam or an analogue thereof be another road to an antidepressant? I notice euphoria from it stronger than tramadol, stronger than orphenadrine, to which nefopam is structurally related, and all three can be mixed and maybe a fourth ingredient (tripelennamine) and a fifth (naproxen) added? I am in the midst of an experiment right now. It's nice.
Would nefopam or an analogue thereof be another road to an antidepressant? I notice euphoria from it stronger than tramadol, stronger than orphenadrine, to which nefopam is structurally related, and all three can be mixed and maybe a fourth ingredient (tripelennamine) and a fifth (naproxen) added? I am in the midst of an experiment right now. It's nice.
checktest
Bluelighter
- Joined
- Jun 9, 2013
- Messages
- 338
The responses some people had to minocycline were interesting as well, in terms of different mechanisms. And of course the rabbit hole of supplements such as methylfolate in addition to SAMe.
As for rubidium, a question could be longer term toxicities like lithium, whether nephrotoxicity or thyroid effects. Well, I guess it might be closer to K than Na and Li, so which channels are relevant could alter that.
Yeah, for SNDRI on other ends guess we'll have to see about some failing development / trials due to more abuse, anorectics and others. At least relative safety, though.
Naproxen is somewhat interesting. I've heard roughly three areas of effect.
1) COX effects. General anti-inflammatory, increased arachidonic acid (shuttled more toward leukotriene path), cytokines, etc... COX-2 inhibition especially, with Celebrex in bipolar depression. (Though aspirin does as well, but maybe that is related to tyrosine hydroxylase or something.)
2) Renal clearance of many drugs, metabolism
3) [Specifically to Naproxen] Relevant GSK3B inhibition. GSK3B was thrown around as possibly a target of lithium, and relevent in bipolar disorde amongst others.
NSAIDs appear to possibly interfere with some antidepressants though, and generally have failed or disappointed. For individuals though it may vary.
As for euphoria and analgesia, there are the interesting people insensitive to pain. Some can feel more pain on naloxone treatment, others don't have that. I'm surprised we haven't seen more development of the sodium channels related to Nav1.7 associated with one of those syndromes. Well I mean selective drug development can be hard, and translating models.
Scopolamine was also trialled as a rapid acting antidepressant in addition to ketamine, with a few comparisons and augmentation protocols between them. Like GluA1 s845 phosphorylation and other similar changes, though scopolamine failed relative to ketamine.
[I won't lie, I cut up a few scopolamine patches during a melancholic episode and tried it. While it was helpful, and my anxiety was far better, scopolamine is a memory impairment model tried and true for a reason.]
As for rubidium, a question could be longer term toxicities like lithium, whether nephrotoxicity or thyroid effects. Well, I guess it might be closer to K than Na and Li, so which channels are relevant could alter that.
Yeah, for SNDRI on other ends guess we'll have to see about some failing development / trials due to more abuse, anorectics and others. At least relative safety, though.
Naproxen is somewhat interesting. I've heard roughly three areas of effect.
1) COX effects. General anti-inflammatory, increased arachidonic acid (shuttled more toward leukotriene path), cytokines, etc... COX-2 inhibition especially, with Celebrex in bipolar depression. (Though aspirin does as well, but maybe that is related to tyrosine hydroxylase or something.)
2) Renal clearance of many drugs, metabolism
3) [Specifically to Naproxen] Relevant GSK3B inhibition. GSK3B was thrown around as possibly a target of lithium, and relevent in bipolar disorde amongst others.
NSAIDs appear to possibly interfere with some antidepressants though, and generally have failed or disappointed. For individuals though it may vary.
As for euphoria and analgesia, there are the interesting people insensitive to pain. Some can feel more pain on naloxone treatment, others don't have that. I'm surprised we haven't seen more development of the sodium channels related to Nav1.7 associated with one of those syndromes. Well I mean selective drug development can be hard, and translating models.
Scopolamine was also trialled as a rapid acting antidepressant in addition to ketamine, with a few comparisons and augmentation protocols between them. Like GluA1 s845 phosphorylation and other similar changes, though scopolamine failed relative to ketamine.
[I won't lie, I cut up a few scopolamine patches during a melancholic episode and tried it. While it was helpful, and my anxiety was far better, scopolamine is a memory impairment model tried and true for a reason.]