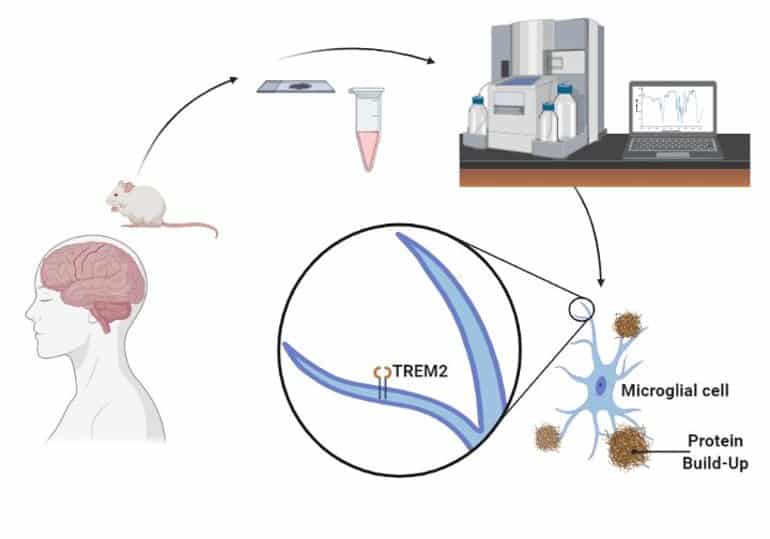
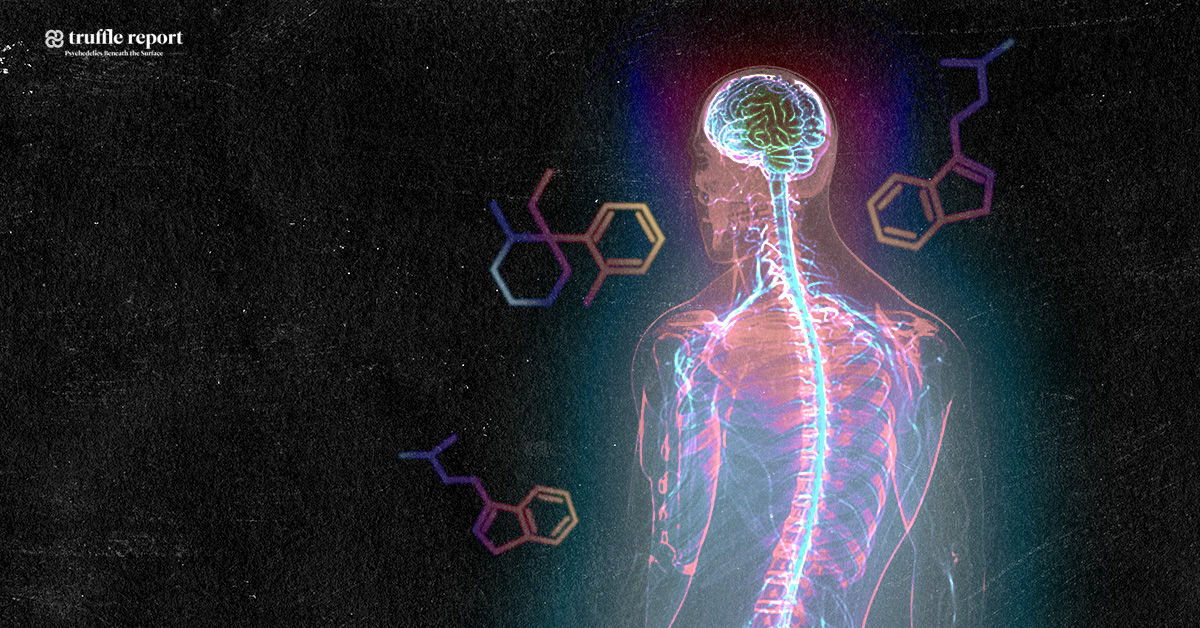
Ayahuasca and the potential treatment of ALS*
by Daniel Gustafsson
This article is the culmination of six years work, having studied ethnobotanical natural medicine and the field of motor neuron disease, making connections between the two in search for viable treatment options for ALS – amyotrophic lateral sclerosis, and similar neurodegenerative conditions.
In south and central america, native people from tribes living within the Amazon rainforest have a long historical tradition of preparing and consuming a natural medicinal tea called ayahuasca. It is harvested and made mainly from a wild growing vine, its latin name being Banisteriopsis Caapi. Often, but not always, leaves from trees named Chacruna and Chaliponga (Psychotria Viridis and Diplopterys Cabrerana) are added to the tea, or in some regions Jurema tree bark (Mimosa Tenuiflora).
The rainforests of the earth are known to be enormously resourceful and vital to the ecosystem of the entire planet. An estimated abundance of undiscovered medicinally valuable plants remain to be explored within these forests, and many current conventional pharmaceuticals originate from substances found in rainforest plants. Ethnopharmacologists are long since aware of widespread support for the medicinal value of ayahuasca and its use in treatment of a wide variety of diseases, but until recently evidence has been limited. This however has changed in the 21st century, as interest and studies in ayahuasca have increased.
Natural substances extracted from ayahuasca plants have been found to possess unique restorative and targeted antioxidative properties on specific nerve cells in the brain and central nervous system, controlling neurotransmission, muscle and motor activity, memory and coordination. This gives probable cause to the theory that ayahuasca could be an effective treatment for neurodegenerative diseases such as ALS, Alzheimer’s, and Parkinson’s disease. Promising results have also been obtained from studying the substance psilocybin, remarkably close in molecular structure to substances found in ayahuasca, naturally occuring in certain species of medicinal mushrooms consumed by the indigenous people where ayahuasca is also used.
According to Dr. Juan Ramos, head of the neurological disease department at the South Florida university, USA, initial studies show that mushroom tryptamine substances semi-identical to the ones contained in ayahuasca stimulate the development of new cells in areas of the brain controlling above mentioned functions. If this could lead to an eventual cure through complete restoration of damaged and lost nerve cells remains to be seen, but initial results indicate this could potentially be the case. Other studies led by Dr. Jordi Riba at the spanish university of Sant Pau, Barcelona, show connections between ayahuasca and neural pathway redevelopment in the neocortex.
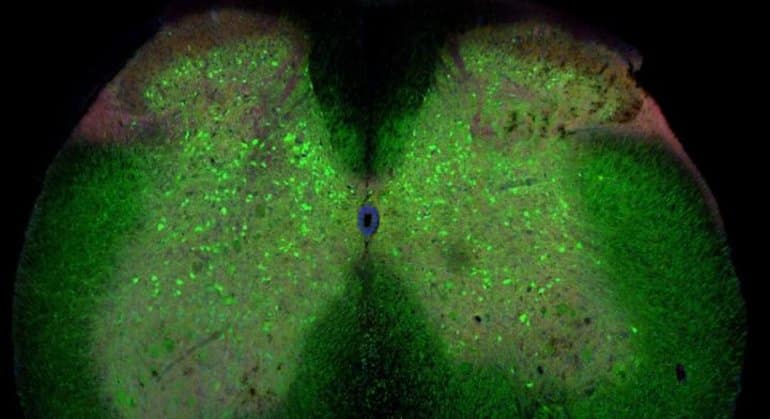
Cancer researchers have also shown interest in B. Caapi, as its alkaloids have shown to be effective against the growth of cancer cells, and are believed to be able to stabilize and balance mitochondrial function. This relates also to ALS research in that mitochondrial dysfunction is nominated one of the main causes of cell damage in ALS, and that the normalization of mitochondrial metabolism through modulation of calcium influx from beneficial alkaloids contained in ayahuasca could prevent motor neuron damage and increase nerve cell survival rate. Mitochondrial function is directly related to neuronal survival, and unregulated levels of intracellular calcium are thought to initiate motor neuron dysfunction, or amplify other mechanisms prone to injure motor neurons.
Eduardo E. Schenberg, Federal University of Sao Paulo:
“There are enough available evidence that the active substances in ayahuasca, especially dimethyltryptamine and harmine, have the positive effect of preventing cancer cells in cultures used for cancer research, and that these substances affect the biochemical processes that are crucial to the treatment of cancer in-vitro as well as in-vivo. The reports available about people with experience from ayahuasca in the treatment of cancer should be taken seriously. The hypothesis is that the combination of (beta-carboline) alkaloids and dimethyltryptamine present in ayahuasca blocks the transportation of nutrients to tumours, lessens the dividing process of cancer cells, and changes the unbalanced mutation-causing metabolism in cancer cells.”
A recent study by Icahn School of Medicine, New York, singled out harmine (from the ayahuasca Caapi plant) among 100.000 substances, as the only one able to cause beta cells in the pancreas (the internal organ that produces insulin) to regenerate, a discovery of great interest to diabetes researchers. Other evidence suggest that ayahuasca may have the potential to regenerate several different types of cells, in many places in the body where needed, the specifics of which calls for medical research in many areas – especially neurodegenerative diseases without a known cure so far. There is also a growing interest in exploring the cell regenerative properties of these plants in spinal cord injury research. Harmine in ayahuasca has also been found to regulate glutamate pump expression in the central nervous system, thereby reducing glutamate toxicity – one of the causes believed to trigger and aggravate ALS through excitotoxic reactions occuring through excessive receptor stimulation by neurotransmitters.
Previously controversial about ayahuasca, was the fact that the plants included used to be thought of simply as “hallucinogenenic” by western science. In other words, ayahuasca and similar plants were neglected by the scientific community and disregarded as if they were nothing more than natural drugs. An objective and more accurate term for this category of plants, in respect to the indigenous culture in which ayahuasca is a part, is the term “entheogenic” – meaning plant that are used in a context sacred to native people, inducing spiritually oriented experiences (according to their own cultural perspective and worldview). In several countries, such as Peru, ayahuasca is fully legal and accepted as a complement to conventional medicine, and during recent decade, western countries have increasingly become more accepting towards entheogens such as ayahuasca, as more studies of entheogenic plants have been completed, changing a formerly unfounded an negative attitude.
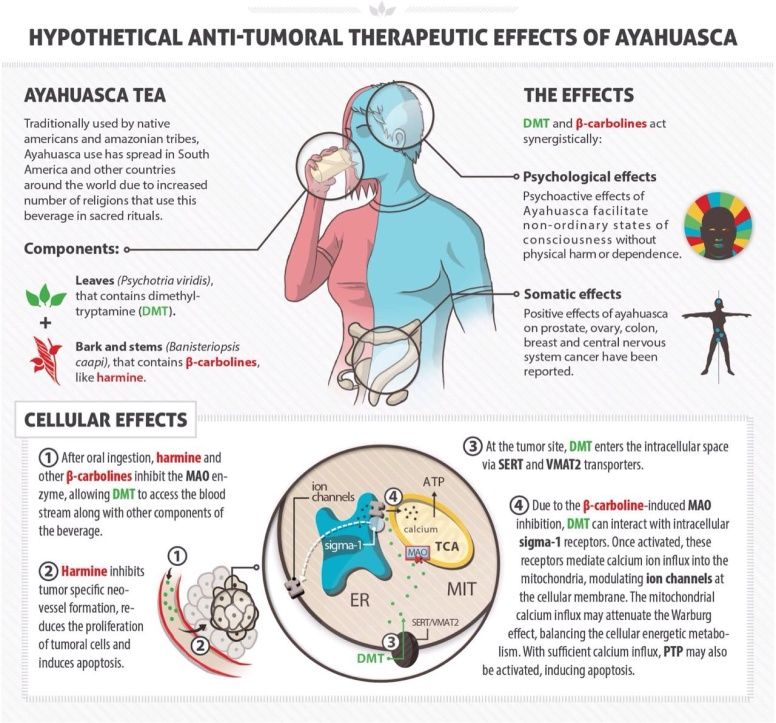
Along several other similar harmala-alkaloids found in B. Caapi, harmaline is a monoamine oxidase inhibitor. Monoamine oxidase (MAO) is an enzyme in the body that breaks down signal substances (such as serotonin). The inhibition of MAO allows the signal substance to remain in the synapse for a longer period of time. Many anti-depressants work in a similar manner, as they stimulate receptors in a targeted area. However, the alkaloids present in ayahuasca should not be compared to antidepressants, as they are not exactly comparable though they both have the ability to affect the same receptors. A comparison is that Caapi alkaloids and antidepressants have the same type of delivery system, but different contents. The biochemical properties of plants used in ayahuasca, and the effects they cause on a multitude of bodily functions remain unique to these plants alone. Various types of harmala alkaloids exert suppression of neurotoxic metabolites, such as quinolinic acid and kynurenine – metabolites correlating with ALS, Alzheimer’s disease, Parkinson’s disease and Huntington’s disease, all in which elevated levels of given metabolites are found and thought to contribute to onset of disease through interaction with spinal motor neurons.
Harmaline together with harmine and tetrahydroharmine,
all forms of mao-inhibitors, are the main components in
the medicinal plant Banisteriopsis Caapi
Ayahuasca in itself is proven unharmful, its compounds being non-toxic, although temporary side effects such as nausea and vertigo are common when used in amounts above medicinal purposes. However, combining certain medical drugs with MAO-inhibitors (such as the ones found in ayahuasca) can be dangerous, even lethal in some cases. This means that in order to safely consume ayahuasca, it must not be combined with any counterindicated pharmaceutical drugs, and those suffering from certain health conditions such as epilepsy or high blood pressure are adviced to either refrain from use or to proceed only with caution and supervision. Uncomfortable side effects from ayahuasca are greatly dose dependent, and a smaller amount consumed for medicinal purpose can thus easily equal few if any side effects experienced.
When searching for information about ayahuasca, a few negative articles can be found, emotionally angled (understandably so), since they tell stories of unfortunate tourists who on their own, or having been duped into doing so, drink something entirely else than ayahuasca – for instance the toxic plant datura, or liquid made from tobacco plant – with serious outcome to their health (including death in a few known cases, from apparent nicotine poisoning). This leads to fear and misinformation, and is not only tragic for the diseased and their families, but also for the natural medicine community trying to promote safe and responsible use of natural medicine for health benefits, and treatment of diseases that regular medical care have failed providing options for. Sensationalistic headlines making unfounded claims, written by people without any knowledge about ethnobotanical medicine, will definitely not help neither ALS patients or people searching for information, and only makes medicinal plant research more difficult. In several countries, including Peru, Brazil and Costa Rica, established retreats offer ayahuasca treatment where the correct plants are harvested (and sometimes organically grown on the property) and prepared by experienced botanists.
One of the earliest studies on B. Caapi were done in the 1920’s, and involved patients with Parkinsons’s disease. The patients experienced great symptom relief in early trials, but unfortunately the research was prematurely discontinued due to lack of profit potential – as substances already present in natural plants could not be applicable for patents useful to pharmaceutical companies.
Ayahuasca therapy is likely to gain further attention in coming years, but is already well established in some places. Should discoveries eventually lead to production of a therapeutic pharmaceutical drug derived from these plants, it would take many years until a final widespread and established product reaches and helps actual patients. The process from studies, through trials, to eventual launch of an approved pharmaceutical made for use in the medical care system, is slow due to obvious reasons. The real and interesting fact is that ayahuasca in its natural form is something already available to people with diagnoses lacking other treatment options. For those who can and want to use ayahuasca, there is, while not in any way guaranteed, a possibility for improvement that is still real. As in many other cases, individual results vary, and there should be an emphasis on not overly promoting this medicine (and thereby people’s hope) while questions and work remain. There is also the importance of emphasizing and thereby minimizing the risks involved concerning contraindications with other medications and conditions. Awaiting further studies, this information should still be worth the attention of those suffering from a debilitating progressive disease such as ALS, and researchers in medical science.
My personal connection to this topic and project, was the passing of a close friends’ mother in ALS which occured in 2012. The course of her disease was rapid and aggressive, and unfortunately several of the now available studies referred to in this article, were not yet published at the time. This led me into investigating biochemical aspects of entheogenic plant medicine in search for treatment of neurological disease, and making information on the topic publicly available, while connecting it to a study currently in progress.
B. Caapi is legally obtainable in most countries and states much in the same way as other known herbal remedies, such as Echinacea and Ginseng. However, as with any other potent natural supplement, it is up to the consumer to use and combine supplements and herbal remedies in an informed and responsible way. In the same way as conventional pharmaceuticals, natural medicines should always be treated with proper respect. One commonly known species that actually contains small amounts of harmala alkaloids is passion flower, or passion fruit tree, although the concentrations in its leaf foliage and flowers are far too low (and the fruit contains none) to be used effectively for monoamine oxidase inhibition and ayahuasca purposes. Also, while similar to some degree, the alkaloid profile in terms of proportions and molecular structural deviations between distinct alkaloids does not match up exactly with that of B. Caapi, making the two species related from a certain aspect, but far from equal regarding their medicinal properties. Extracts from various passiflora species are however produced and sold worldwide as mild herbal relaxants and sleeping aids, and as an antispasmodic for treating Parkinson’s.
The substance known as dimethyltryptamine, found in plants traditionally added to ayahuasca, is regulated by law in a number of countries classified as a scheduled substance. (Questionably so, due to medicinal value in multiple areas). This particular substance contained in the additional aforementioned plants induces the altered state of consciousness, a many times misunderstood and stigmatized phenomenon. A description of this altered state is that it is dreamlike, that it stimulates memory and the ability to think abstract, and has self-therapeutic qualities. Even though dimethyltryptamine is naturally occuring in the human body, thought to be produced in small amounts by the pineal gland in the brain during sleep, it remains an illegal substance in a number of western countries since the 1960’s and 70’s, when lawmakers prematurely criminalized almost any substances suspected of having any effect on the mind, including natural ones, due to the widespread moral panic at the time – regardless of the fact that many of them, including dimethyltryptamine, were never proven unhealthy, having in fact been used by indigenous people, in the form extracted from plants, to successfully treat diseases and health problems for centuries. Leaf juice from Chacruna has been used traditionally as a remedy for migraines and ant bites, and Jurema bark for treating burn wounds – significantly quickening regeneration of skin and scar tissue. Dimethyltryptamine also targets chaperone sigma 1, a receptor subtype expressed in both neurons and glia of multiple regions within the central nervous system, with capacity to modulate biological mechanisms implicated with neurodegeneration. Sigma 1-receptors present compelling targets for pharmacologically treating neurodegenerative disorders, and dimethyltryptamine acts as an endogenous Sigma-1 receptor regulator, but interactions between the two in association with motor neuron disease is not understood.
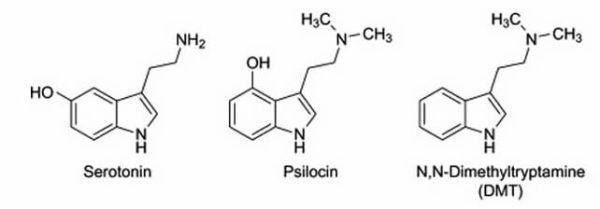
Comparison between Dimethyltryptamine, Psilocin- and Serotonin-molecules
(Psilocybin being the precursor to Psilocin)
Although, several european countries have redefined their policy regarding many formerly frowned upon medicinal plants in recent years, much due to an increasing awareness and access to new and unbiased information and up-to-date research regarding these plants. In Scandinavia, Sami natives from Sapmi, Sweden, were recently aquitted from all charges in the court of law, for having brought Peruvian medicinal cactus into the country. The court established that natural plant material alone cannot be defined as a scheduled substance, and that the therapeutic work involved people were doing, which included Echinopsis Pachanoi cactus, was indeed not a criminal act, but served the purpose to help and heal people. Another similar case with the same outcome involved ayahuasca additive plants. Cacti from the Echinopsis and Lophophora species are known for their restorative and cleansing effects on the body, and are used as such in ethnobotanical medicine.
To be precise, the definition of Ayahuasca is any tea made from either the plant Banisteriopsis Caapi alone, or from B. Caapi and additional plants containing dimethyltryptamine. A tea made from B. Caapi alone does not have what is sometimes referred to as “visionary” qualities, as only the addition of dimethyltryptamine from additive plants, or actually the combination of mao-inhibiting alkaloids in B. Caapi together with dimethyltryptamine content induces a state of mind formerly mislabeled “hallucinogenic”. It needs to be clarified that this word brings up negative associations in many people, and is thus feared and misunderstood. Unlike what some people tend to think, one does not hallucinate things appearing out of thin air after having consumed ayahuasca, but rather there are sequences of inner dreamlike visions taking place while resting, while still awake and fully conscious, provided a significant amount of tea has been consumed. It is actually quite undramatic, aside from the unpleasant vomiting which does affect some people (which is entirely intentional in traditional in cleansing rituals).
And herein lies the essence many times misunderstood: It does not take a great amount of ayahuasca to experience strictly its medicinal effects – without abstractions and visionary effects, or nausea/vomiting which naturally people want to avoid. Several medicinal health benefits can be obtained even by using B. Caapi alone – without additive plants, thereby ensuring no visual effects experienced at all, should this be desired. It should be noted though, that the synergistic effect between the two plants used simultaneously will bring the best medicinal and bodily response. Exaggerations regarding ayahuasca made these medicinal plants overlooked for many years in the west, but the reputation has been revised as more people with experience in a medicinal context have made statements, claiming the medicinal value of ayahuasca relevant to medical conditions of different types – the field of neurological disease being the latest. Ayahuasca has already been effectively used for symptom relief from Multiple Sclerosis and rheumatoid arthritis, by a growing number of people in Europe since at least 2006. ALS, Multiple Sclerosis, Alzheimer’s and Parkinson’s disease all share a lot of common ground, being that they all involve varieties of nerve cell degeneration. Thus it is likely that any type of natural broad spectrum medicine able to affect the process of nerve cell regeneration, and that also has substantially antioxidative and cell protective properties, could prevent and slow the progression of neurological disease in general.
Whatever wild or strange stories about ayahuasca that may occasionally be found circulating, they stem mostly from people who went to live with native tribes during the late 80’s and early 90’s, taking part in traditional ceremonial use of ayahuasca – consuming exceptionally generous or concentrated amounts of the medicine, enfolding themselves in deep cleansing experiences not necessarily easily endured. This medicine, like all others, should definitely be well respected, but not subject to exaggeration or downright misrepresentation – causing people to dismiss what they are simply uneducated about, which in turn may lead to people never getting treatment that could slow the progression of their terminal health condition, or in some cases even reverse it. The vivid and fascinating visions induced by strong tea often seem to have a theme rooted in nature, perhaps tied into the underlying psychological expectations associated with the revered history of ayahuasca itself, as depicted quite beautifully in colorful paintings by Peruvian artist Pablo Amaringo (1938-2009). The visions arise from the simple fact that the alkaloids and tryptamines dissolved in the tea, combine to affect receptors that in turn stimulate the processing of memory relating to images and words – noticeably of relevance to Alzheimer’s research.
Ayahuasca is proven non-addictive, and is even used to aid people in breaking their drug dependencies, as ayahuasca has a detoxifying and documented effect of ridding the user of drug related abstinence issues.
MAO-inhibition makes uptake of dimethyltryptamine in the body possible, unable to occur otherwise as dimethyltryptamine without MAO-inhibition is rapidly broken down by enzymes in the stomach, unable to cause effect. Dimethyltryptamine is almost molecularly identical to the above mentioned psilocybin from Dr. Ramos research. Researchers think that psilocybin and dimethyltryptamine binds to brain receptors that stimulate growth and healing, acting on the hippocampus, a part of the brain that is essential to learning and forming memories, recieving sensory impulses and that has target cells and receptors for important signal substances. Hippocampus atrophy occurs when nerve cells die, or when abnormal levels of stress hormones prevent neurogenesis, and is a known sign in Parkinson’s disease. It is theorized that the unique combination of a variety of harmala-alkaloids from B. Caapi, and dimethyltryptamine from additional plant sources used in ayahuasca, work on cellular level to repair and restore nerve cells, stimulate and enhance motor neuron transmission, and to protect remaining nerve cells from degeneration. This is without doubt valuable from a neuromedical perspective.

Ayahuasca plants as packaged and sold in health food stores in, Peru.
As the non regulated B. Caapi alone has proven positive abilities, potentially effective against neurological and cancer diseases, it is thus something real and available that may be a valid treatment option. For anyone who experience positive results to any degree, but does not live in a state or country where the use of plants containing dimethyltryptamine is permitted, there is the possibility to travel to one of the many countries (or states) which by law allows the use of added secondary plants with their combined medicinal properties for evalution of full-spectrum ayahuasca treatment. In Europe, Spain is one of several countries where ayahuasca is becoming an established therapy, and Spain is also the chosen location for an international conference 2014, where ethnopharmacologists, psychologists and researchers from all over the world gather around the topics of ayahuasca and medicinal entheogens.
Among others, Ede Frecska, M.D., Ph.D, University of Debrecen, lectures on the possibilities of recreating braincells and regulating the immune defense system through this plant-based medicine and others. This event is held by ICEERS – International Center for Ethnobotanical Education Research and Service, and can be followed at:
Examination of Caapi medicinal vine specimen.
Furthermore, besides ability to aid and enhance the process of nerve cellular repair and protection against cell oxidation, several entheogenic plants (and fungi), including ayahuasca, do possess psychotherapeutical qualities as well. Coping with degenerative illness is obviously stressful to patients, and a great deal of emotional relief, personal insight, and ability to better cope with personal situation is achievable through the single or repeated experience of entheogenic medicinal plants/mushrooms in a comfortable and supportive environment, according to renowned Johns Hopkins medical university.
The fact that many of these medicinal plants are becoming revived as they recieve scientific approval, is great news in several ways. Sustainability and environmental issues comes to mind, and so far the outlook remains positive. Many organic farms have developed in south and central america, cultivating ayahuasca plants for both local use and for export, providing work and income for people in rural areas otherwise stricken with poverty. This also serves as a way for numerous local people to reconnect with their cultural past, as ayahuasca is declared a national heritage in Peru among other places.

Dennis Mckenna, PH.D, one of the world’s most renowned ethnopharmacology researchers speaks on the importance of sustainability, regarding cultivation of ayahuasca and the preservation of rainforests. This should concern all people who benefit from treatment using ayahuasca medicinal plants. Dr. Mckenna is co-founder and director of ethnopharmachology at Heffter Research Institute, New Mexico. He is also a faculty member of the Academic Health Center at the University of Minnesota, and was key organizer in the Hoasca Project, an international biomedical study of ayahuasca, funded by the Stanley Medical Research Institute. While ayahuasca plants take years to mature for harvest, they can actually be grown at home in gardens anywhere in the world where the climate allows, or indoors or in heated greenhouses elsewhere, and the seeds are both cheap and abundant – available from numerous online ethnobotanical vendors worldwide, ensuring the survival of these medicinal plant species and their sustained availability for future generations.
Formerly unknown to science, we are beginning to understand the potential and medicinal aspects of this particular plant, replacing neglect of entheogens with knowledge as scientific groundwork in this matter becomes firm.
People should not be led into thinking this is some kind of natural miracle cure, but used the right way it could provide longterm aid in the restorement of body and mind function in certain neurological conditions. Together we can all inform people in an unbiased, ethical and safe way about any and all viable treatment options, and about the medicinal and therapeutical value of entheogens in general.
Common sense should be applied to determine dosage in order to ensure a sufficient degree of medicinal activity, while not being harmful or overwhelming. Specific diet is essential in conjunction with ayahuasca, in order to minimize side effects and to maximize utilization of medicinal compounds, and basic knowledge on the process of preparation of the medicine is helpful in order for the plants to synergize correctly. The help and information needed for these purposes, along with protocols for detoxification are individually provided to participants in the below pilot study.
Ayahuasca has been used for a very long time historically, and only recently for treatment of the conditions brought up in this article. Any substantial health improvement from this natural medicine in cases of motor neuron disease would be likely to reveal itself long-term at first. Initial updates from people participating in the ayahuasca ALS treatment pilot study report a few things in common; wider range of movement, weight gain in muscle mass, muscle tension relief, reduced spasticity and improved strength in affected limbs, though it should be noted that none of the participants had lost all muscle control prior to treatment, and that whether or not this effect turns out to be persistent or permanent is not known at the moment.
Summary of key points:
Ayahuasca could effectively be used in treatment of ALS and other motor neuron diseases based on the fact that studies suggest uniquely antioxidative effects that seem to protect brain/nerve cells, targeting motor neurons through a unique biochemical transport system, and that it and other moleculary similar substances, also naturally occuring, stimulate neurogenesis – the development of new brain/nerve cells, and improved neural connectivity. In studies it has been found to reduce symptoms in Parkinsons’s patients – all neurodegenerative diseases share common ground, making it likely that something that improves one major neurological condition could also be beneficial to other closely related conditions. Also based on credible personal accounts from people having used ayahuasca for symptom relief from their multiple sclerosis (once again – the common ground of neurodegenerative diseases), documented in books about ayahuasca, and descriptions of early stage minor improvement by Ayahuasca ALS Treatment study participants, already having used the medicine for a period of time. Studies also indicate ability to normalize metabolism in mitochondria crucial to motor neuron survival, and to regulate and decrease levels of excitotoxicity in the central nervous system.
Ayahuasca and other entheogens can and will gain the credibility and amends they truly deserve, and bring new possibilities to many people living with diseases lacking clinical healthcare treatment options. Until then, these medicinal plants remain available for personal evaluation by individuals who choose to explore the option. In relation to the medical conditions brought up in this article, these plants may hold future keys in becoming a powerful tool for reversal of ALS progression and related conditions. Share this important information if you find it valuable.
*From the article here :
By Daniel Gustafsson Swedish version/Artikel på svenska: This article is the culmination of six years work, having studied ethnobotanical natural medicine and the field of motor neuron disease, mak…

ayahuascatreatment.wordpress.com