4. Receptors
Receptors are categorized by their structure and different mechanisms of action. Classification can differ depending on the book you look it up so here's the one I like the most:
1. Ionotropic receptors are kind of the simplest ones in that there's no second messenger or DNA transcription involved. They're trans-membrane receptors and can be thought of as a channel going through the cell membrane. When a agonist (i.e. drug) binds to it, the receptor will change conformation allowing ions to flow through inside or outside the cell, depending on the ion. That's why they're called IONotropic. The GABA-A or glycine receptor would be examples.
2. Metabotropic receptors are cell-membrane receptors too, but the difference to ionotropic receptors is that they lead to a signal-cascade on the intracellular side of the cell. They can be divided further into GCPRs as the most important ones and other types like tyrosine kinase receptors and others, but those aren't that important for understanding drugs of abuse.
2.1 G protein coupled receptors (GPCR). GPCRs don't have a channel for ions, but as the name suggests, these are receptors that are coupled to another protein, called G-protein, consisting of the alpha, beta and gamma subunit. When a ligand binds to the receptor the receptor in turn activates the G-protein. The G-protein now dissociates into the alpha subunit and the beta-gamma subunit. The alpha unit now activates/deactivates (depending on which type it is) another protein downstream. There are a few alpha units i.e. Gs, Gi, Gq/11 and G12/13. Examples for GPCRs are the adrenoceptors, muscarinic acetylcholine receptor, opioid receptors and dopamine receptors.
For the sake of completeness there are protein synthesis regulating receptors and toll like receptors too, but those aren't important for understanding the mechanism of most drugs of abuse. Additionally there's something called biased agonism or functional selectivity, meaning that a drug can selectively activate/deactivate a certain signal pathway (for example 2C-N binds to 5-HT2B but only leads to the activation of the second messenger PLA2 without activating PLC, this leads to the release of arachidonic acid without the production of IP3) but afaik this is still not fully understood and would be too much for this thread.
4.1 GABA receptors
4.1.1 GABA-A
-GABAA-receptor-protein-image
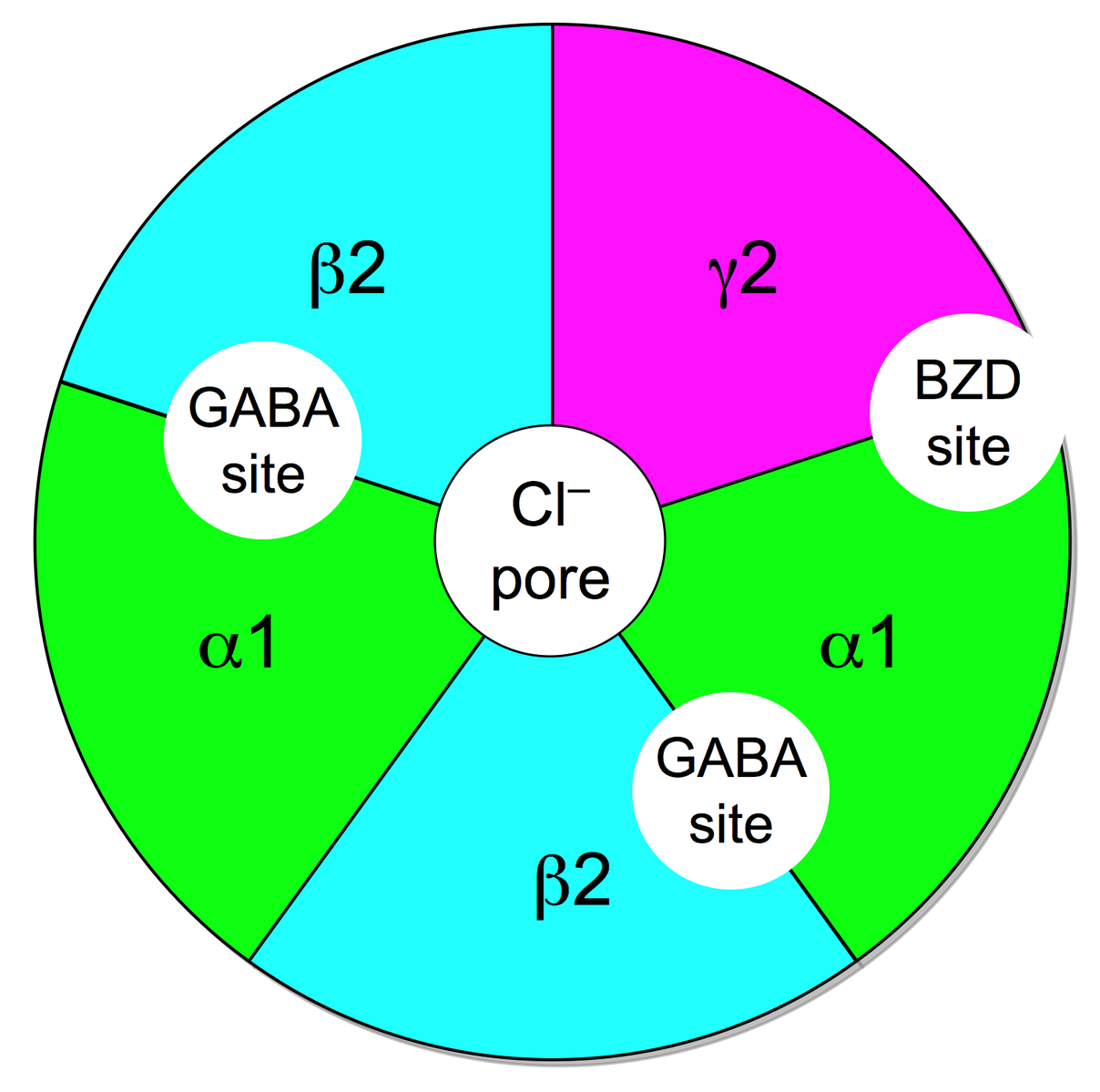
The GABA-A receptor belongs to the ligand gated ion channels and selectively conducts chloride-ions, leading to an inhibitory postsynaptic potential, meaning that the postsynaptic neuron is less likely to generate an action potential, overall leading to inhibitory effects.
The receptor is a pentamer, meaning that it consists of five subunits. The subunits can be split into various isoforms that are important for properties like agonist affinity etc. The alpha unit can be divided into alpha 1-6, with alpha1 being important for the sedative effects of benzodiazepines, alpha2 for the anxiolytic effects and alpha3 for the muscle-relaxant properties. Then there are three different beta units as well as three gamma units. And finally each one delta, epsilon and theta unit. Most commonly a GABA-A receptor consists of two alpha, two beta and one gamma unit.
There are various endogenous and exogenous ligands with different binding sites. There's the orthosteric binding site (that's where the endogenous ligand GABA binds to) and different allosteric sites (a binding site where the endogenous ligand doesn't bind to).
To activate the receptor two GABA molecules have to bind to the GABA binding site located between the alpha and beta unit. The activation usually leads to an influx of chloride-ions.
Benzodiazepines bind to a allosteric binding site located between an alpha and gamma subunit. For the binding to happen the alpha unit has to contain a histidine residue.
Other binding sites include the loreclezol binding site (valerenic acid most likely binds to this site) and possibly a binding site for ethanol and different volatile anesthetics.
4.1.2 GABA-B
GABA-B is, like GABA-A, a mostly inhibitory receptor, but unlike GABA-A it belongs to the GPCR-family. GABA-B is Gi-coupled and leads to postsynaptic activation of potassium channels and presynaptic inhibition of calcium channels (mostly N-type or P/Q-type). Those effects cause an IPSP and thus decrease the excitability of the downstream neuron.
This receptor is distributed throughout the central and peripheral nervous system, with the highest density in the cerebral cortex, thalamus, cerebellum and amygdala.
Drugs that bind to GABA-B include ethanol, Baclofen, GHB in high doses and Phenibut.
4.2 Opioid receptors
The opioid receptors include the mu-, delta- and kappa-receptor, all of which modulate pain perception. All three are GCPR located in the brain, spinal cord as well as in the gut and bladder. They act pre- as well as post-synaptic. Presynaptic the Gi protein decreases cAMP via a second messenger and causes calcium channels to close, effectively inhibiting the release of substance P and glutamate. Postsynaptic activation of opioid receptors leads to the opening of potassium channels resulting in the hyperpolarization of the postsynapse reducing neurotransmission.
The mu-receptor can be subdivided into mu-1 and mu-2, both of which are the target of most pain meds on the market. Mu-1 is implicated in analgesia, sedation, euphoria, miosis, cough suppression and physical dependence. Mu-2 additionally leads to respiratory depression and constipation.
The delta receptor is implicated in analgesia, antidepressant effects but also in physical dependence and convulsant effects.
The kappa receptor is implicated in analgesia, miosis, sedation, neuroprotection and anticonvulsant effects, but side effects are depression, diuresis and hallucinations (
Salvinorin A is a selective kappa-receptor agonist).
4.3 Adrenoceptors
The adrenoceptors are a class of GCPR that are subdivided into
alpha- and beta-adrenoceptors. They are targets of many catecholamines like epinephrine and norepinephrine as well as many drugs.
Activation generally leads to stimulation of the sympathetic nervous system and thus is responsible for the fight-flight-or-freeze response leading to an increase in heart rate, increase of blood flow into skeletal muscle and dilated pupils.
Alpha-1 is Gq/11-coupled and activation causes mostly smooth muscle contraction via PLC induced cleavage of PIP2 into DAG and IP3 which in turn increases intracellular calcium. This leads to vasoconstrictions of blood vessels of the skin, GI-tract, kidneys and brain as well as contraction of the urinary bladder. In the brain alpha-1 stimulation produces anorexia and partially mediates the appetite suppressant effects of amphetamine.
Norepinephrine has higher affinity for alpha-1 than epinephrine.
Alpha-2 couples with Gi and is mostly located on vascular prejunctional terminals, inhibiting the release of norepinephrine in a form of negative feedback. In addition alpha-2 receptors are located on vascular smooth muscle cells of certain blood vessels, like skin arterioles and veins. Activation causes Gi to dissociate from the complex and inactivate adenylyl cyclase decreasing cAMP levels. Agonists are have sedative, muscle relaxant and analgesic effects.
Norepinephrine has slightly higher affinity for alpha-1 than epinephrine.
Beta-1 is Gs-coupled and is expressed predominantly in cardiac tissue. Activation leads to a cascade of effects that increase the availability of calcium in cardiac myocytes. Stimulation results in positive chronotropic, dromotropic and inotropic as well as positive lusitropic effects.
Affinities: Isoprenaline > Epinephrine > Norepinephrine
Beta-2 is directly associated with L-type calcium channels and coupled to Gs (and can couple with Gi as well). Agonism leads to adenylyl cyclase activation which in turn increases cAMP concentration triggering PLA which in turn phosphorylates MLC-K causing smooth muscle relaxation. This in turn leads to a decreased motility of the gut, bronchodilatation, increased blood flow to skeletal muscle and as beta-2 activation has stronger effects than alpha-1 on the detrusor urinae muscle urinary retention.
Affinities: Isoprenaline > Epinephrine > Norepinephrine
Beta-3 receptors are Gs-coupled and mainly located in adipose tissue and agonism leads to increased lipolysis.
Affinities: Isoprenaline > Epinephrine > Norepinephrine
4.4 Dopamine receptors
The dopamine receptors consist of 5 different receptors,D1-5, categorized in D1-like (D1 and D5) and D2-like (D2, D3 and D4), all of which are GPCR, but the coupled G-protein varies.
D1-like are Gs coupled (D1 additionally coupled with Golf), thus activation stimulates adenylyl cyclase leading to the rise of cAMP increasing PKA activity.
D2-like are Gi coupled, thus inhibiting adenylyl cyclase, decreaseing cAMP concentration ultimately decreasing PKA activity. Additionally activation leads to opening of potassium channels.
Evidence suggests that G-proteins isn't the only mechanism involved in signaling.
D1 receptors are located on smooth muscle cells of the kidney and mesentery vessels (activation leads to vasodilatation thus better perfusion of kidney and gut) as well as high levels in the basal ganglia (dorsal striatum and ventral striatum, important for motor function as well as learing, cognition and emotion).
D2 receptors are located in the nucleus accumbens, corpus striatum, area postrema (nausea) and anterior pituitary (inhibits release of prolactin). Alternative splicing results in D2Sh (short, presynaptic autoreceptor) and D2Lh (long, postsynaptic).
D2 receptors seem to be down regulated after addictive stimuli like cocaine, heroin or alcohol.
Hyperstimulation of D2 receptors in the mesolimbic system lead to positive symptoms of schizophrenia, while hypostimulation of D2 receptors in the mesocortical system lead to negative symptoms.
D3 receptors are located, among others, in the cerebellum, ventral striatum and nucleus accumbens.
D4 receptors are located in the hippocampus, amygdala and frontal lobe. Mutations/polymorphisms of the gene coding for the D4 receptor have been associated with various behavioral phenotypes, including novelty seeking, ADHD and schizophrenia.
D5 receptors are located in the kidney (regulating sodium excretion), dentritic and T helper cells and in the CNS for example in the hippocampus and amygdala. D4 receptors show 10 times higher affinity for dopamine than D1 receptors.
Dopamine signaling involves many different pathways, the four most important ones are:
a) Mesolimbic (reward-related and aversion-related cognition);
b) mesocortical (executive functions);
c) nigrostriatal (motor function, with substantia nigra); and
d) tuberoinfundibular (inhibition of prolactin release) pathway and hypothalamospinal (motor function) projection.
I'll try to go into more detail in the upcoming chapter 7.
If you're really interested in the dopamine receptors you can check out this book:
'The physiology, signaling and pharmacology of dopamine receptors'
http://pharmrev.aspetjournals.org/content/63/1/182
4.5 Serotonin receptors
Serotonin receptors (5-HT receptors) are located, among others, in the CNS, gastrointestinal tract (here's most of the serotonin), cardiovascular system and platelets. With the exception of 5-HT3 (ligand gated ion channel) are all GPCRs.
5-HT1 has 5 characterized subtypes: A,B,D,E,F (after 5-HT1C was cloned and characterized it seemed to have more in common with 5-HT2 than 5-HT1 so it was redesignated as 5-HT2C)
5-HT1 is Gi coupled so activation leads to inhibition of adenyly cyclase thus reducting cAMP levels
5-HT1A is located in blood vessels and the CNS and is implicated in, among others, anxiety (people suffering from anxiety and depression seem to have reduced density of this receptor in certain parts of the brain) and memory as well as regulation of temperature and blood pressure
5-HT1B and 5-HT1D are Gi coupled as well and are located in the CNS and blood vessels. They play an important role in migraines, as they are located in meningeal blood vessels. Activation of postsynaptic 5-HT1B leads to vasoconstriction while activation of presynaptic 5-HT1D leads to inhibition of neuropeptide secretion.
5-HT1E is located in both the CNS and blood vessels
5-HT1F is found in the CNS and seems to be important for suppression of neuronal inflammation.
Migraine hypothesis: Activation of 5-HT2B leads to NO release which in turn causes vasodilatation in meningeal blood vessels and stimulation of the trigeminal nerve. This leads to the secretion of neuropeptides which reinforce the vasodilatation leading to edema and triggers action potentials that cause nausea and pain
5-HT2 has 3 characterized subtypes: A, B and C that are Gq coupled .
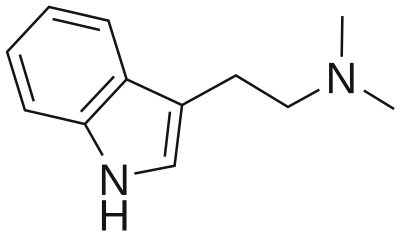
5-HT2A is pretty much ubiquitous as it's located in blood vessels, CNS, PNS, GI tract, smooth muscles and platelets. The receptor plays important role in hemostasis in platelets and due to vasoconstriction. In the CNS the receptor seems to mediate the effects of classic psychedelics like LSD and psilocin. 5-HT neurons in the brainstem project to glutamergic pyramidal cells in deep cortical layers V and VI and cortical pyramidal neurons. Activation of 5-HT2A receptors located on the glutamergic pyramidal cells leads to the release of glutamate into the synaptic cleft stimulating NMDA and AMPA receptors on cortical pyramidal neurons. In addition 5-HT2A receptors are located on cortical pyramidal neurons as well. Both lead to the increased expression of BDNF (brain-derived neurotrophic factor).
Drugs that target 5-HT2A are used as platelet aggregation inhibitors and to treat high blood pressure. 5-HT2A is also targeted by atypical antipsychotics like clozapine and antidepressants seem to down regulate postsynaptic 5-HT2A receptors leading to the anxiolytic effects.
5-HT2B is implicated in high blood pressure and chronic activation might cause damage of heart valves as it plays a role in the regulation of cardiac structure and function leading to proliferation of cardiac valves fibroblasts
5-HT2C is implicated in sexual behavior as well as food intake (activation leads to the activation of the proopiomelanocortin-system in the hypothalamus as well as decreasing the release of neuropeptide Y and agouti-related peptide, suppressing appetite).
5-HT3 is the only ligand gated ion channel (selective for sodium and potassium) located in CNS, PNS and GI tract with an important role in the Area postrema and afferent vagus neurons. Stimulation leads to nausea.
5-HT4 is Gs coupled and located in the GI tracts, CNS and PNS. This receptor plays an important role in regulating gut motility and heart rhythm.
'Higher' 5-HT-receptors include 5-HT5A (possible regulation of circadian rhythm) and B (pseudogene), 5-HT6 (possibly implicated in learning) and 5-HT7 (regulation of circadian rhythm and blood pressure as well as thermoregulation).
4.6 NMDA receptor
NMDA stands for
N-methyl-D-aspartate-receptor, named after N-methyl-D-aspartate, a selective agonist of this receptor. The NMDA receptor is an ionotropic glutamate receptor, both ligand gated (activated with co-binding of glutamate and glycine/D-serin) and voltage-dependent due to a magnesium/zinc block that only dislodges after AMPA receptor activation depolarizes the cell enough. Activation leads to an influx of mainly calcium as well as sodium influx and potassium efflux. Compared with AMPA receptors NMDA's postsynaptic current last for a few hundred microseconds, which is rather long. Overactivation with a high level of calcium influx is thought to be excitotoxic.
The NMDA receptor is a heterotetramer complex consisting of two obligatory GluN1 subunits and two variable subunits (GluN2 A-D or GluN3 A-B).
NMDA receptors are implicated in learning and memory formation as well as synaptic plasticity.
If you're really interested in the NMDA receptor you can check out this book:
'Biology of the NMDA Receptor'
https://www.ncbi.nlm.nih.gov/books/NBK5283/
4.7 hERG
hERG stands for human Ether-a-go-go Related Gene, a gene coding for the alpha subunit of a cardiac voltage gated potassium channel (called Ikr) responsible for the electrical activity coordinating the heartbeat. Now as you might imagine drugs interacting with this channel are able to cause arrhythmia and in the worst case can kill you. To go a little more into detail, this channel is partially responsible for the repolarization due to potassium efflux. Now if you change the timing of repolarization you get either a too fast (short QT syndrome) or not fast enough (long QT syndrome) repolarization. Both of those syndromes can lead to abnormal heart rhythms and can lead to sudden cardiac death. The long QT syndrome is the more common one and there are quite a few drugs that can cause it, including antihistamines like diphenhydramine, methadone, various antidepressants, cocaine, Dextromethorphan and loperamide. Normally the long qt syndrome only persists as long as you take the drug, but some people are genetically predisposed and the prolongation won't resolve itself after cessation of the drug.
Symptoms of QT prolongation include palpitations, chest pain, fainting (= syncope) and fatigue. This syndrome can be lethal as it can lead to an abnormal heart rhythm called 'Torsade de pointes' which in turn can lead to sudden cardiac arrest. Because of that high doses of drugs that are known to cause QT prolongation and combinations of those drugs can be dangerous and shouldn't be done.
4.8 TAAR1
TAAR1 stands for trace amine-associated receptor 1. TAAR1 is a GPCR coupled with Gs and Gq-proteins and is located in the CNS (presynaptic plasma membrane of monoamine neurons) and peripheral cells/organs (stomach, gut, white blood cells, kidney, lungs). TAAR1 is one of six trace amine-associated receptors and plays an important role in regulating monoamine neurotransmission.
As the name suggests TAAR1's endogenous ligands are trace amines with the following potencies:
tyramine (related to dopamine) > beta-phenethylamine (related to dopamine) > dopamine () = octopamine (related to norepinephrine).
TAAR1 is an intracellular receptor, thus ligands have to diffuse across the presynaptic membrane (methamphetamine) or through membrane transporters ((meth-)amphetamine) to bind to it.
There's some selectivity as for example a drug that is only able to enter the presynapse via SERT will mostly bind to TAAR1 receptors found in serotonin neurons.
With TAAR1 coupled with Gs activation leads to activation of adenylyl cyclase increasing cAMP in the presynapse which in turn can up-regulate the expression of trace amines. Another mechanism is DAT phosphorylation by either PKA (leads to DAT internalization) or PKC (reveres transporter function).
4.9 VMAT (transporter)
VMAT stands for vesicular monoamine transporter. VMAT2 is a protein located in the membrane of cytoplasmic vesicles and is only found in vertebrata. Besides the CNS VMAT2 is located in pancreatic islets (regulates insulin response to glucose) and uterus. Mouse studies suggest a difference of neuronal VMAT2 activity in genders.
In the CNS it's responsible for the transport of the monoamines dopamine, serotonin, histamine and norepinephrine (and sometimes GABA) from the cell cytosol into synaptic vesicles. As antiport VMAT2 swaps intravesicular H+ for monoamines from the cell cytosol.
VMAT2 possesses at least two binding sites characterized by tetrabenazine (for the treatment of hyperkinetic movement disorders, for example seen in Huntington's disease, mechanism not fully understood but most likely reversible depletion of monoamines due to VMAT2 antagonism) and reserpine (has been used to treat high blood pressure and psychotic symptoms, works by depleting monoamines). Amphetamines work similarly by inhibiting VMAT2, but, unlike reseprine and tetrabenazine, reverse the direction of monoamine transporters so the cytosolic monoamines can be transported to the synaptic cleft.
Fun fact: in the book 'The God Gene: How faith is hardwired into our genes' by molecular biologist Dean Hamer it's suggested that VMAT2 is responsible for spirituality/'is the gene where God may reside' but the study the claims are based on hasn't been published in a peer review journal and is highly controversial.


")

")