

Deleted member 576750
Discord Sr. Moderator
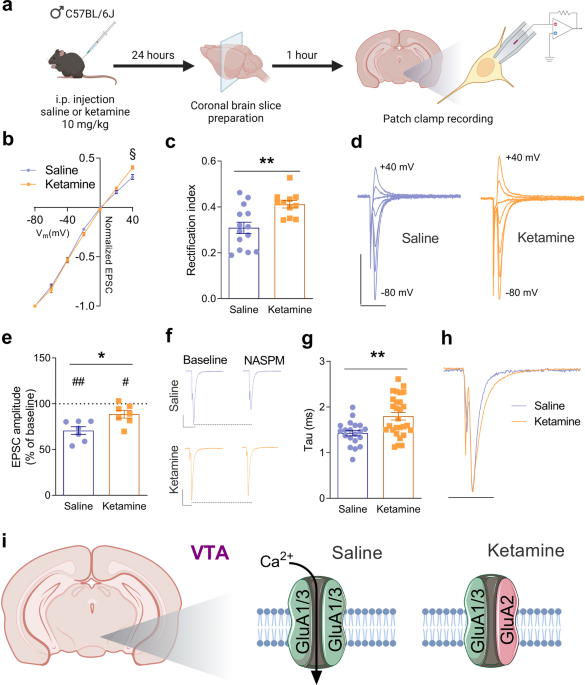
So I've been doing some reading on ketamine recently and came across some interesting info. It sounds like ketamime use may cause some changes in the regulation of AMPA receptors, these changes seem to include upregulation and a change in subtypes leading to greater calcium permeability in some circuits.
Not only is sufficient AMPA activation needed to unblock the NMDA pore, which leads to LTP, but there are also calcium induced LTP pathways through CaMKII (iirc)
Might ketamine use leading to increased rates of LTP at some synapses possibly leading to improved learning in some areas? Food for thought
Maybe I'm just trying to make myself feel better about all the ketamine I've been railing recently
 www.nature.com
www.nature.com
Not only is sufficient AMPA activation needed to unblock the NMDA pore, which leads to LTP, but there are also calcium induced LTP pathways through CaMKII (iirc)
Might ketamine use leading to increased rates of LTP at some synapses possibly leading to improved learning in some areas? Food for thought
Maybe I'm just trying to make myself feel better about all the ketamine I've been railing recently
Ketamine induces opposite changes in AMPA receptor calcium permeability in the ventral tegmental area and nucleus accumbens - Translational Psychiatry
Ketamine elicits rapid and durable antidepressant actions in treatment-resistant patients with mood disorders such as major depressive disorder and bipolar depression. The mechanisms might involve the induction of metaplasticity in brain regions associated with reward-related behaviors, mood...
www.nature.com


