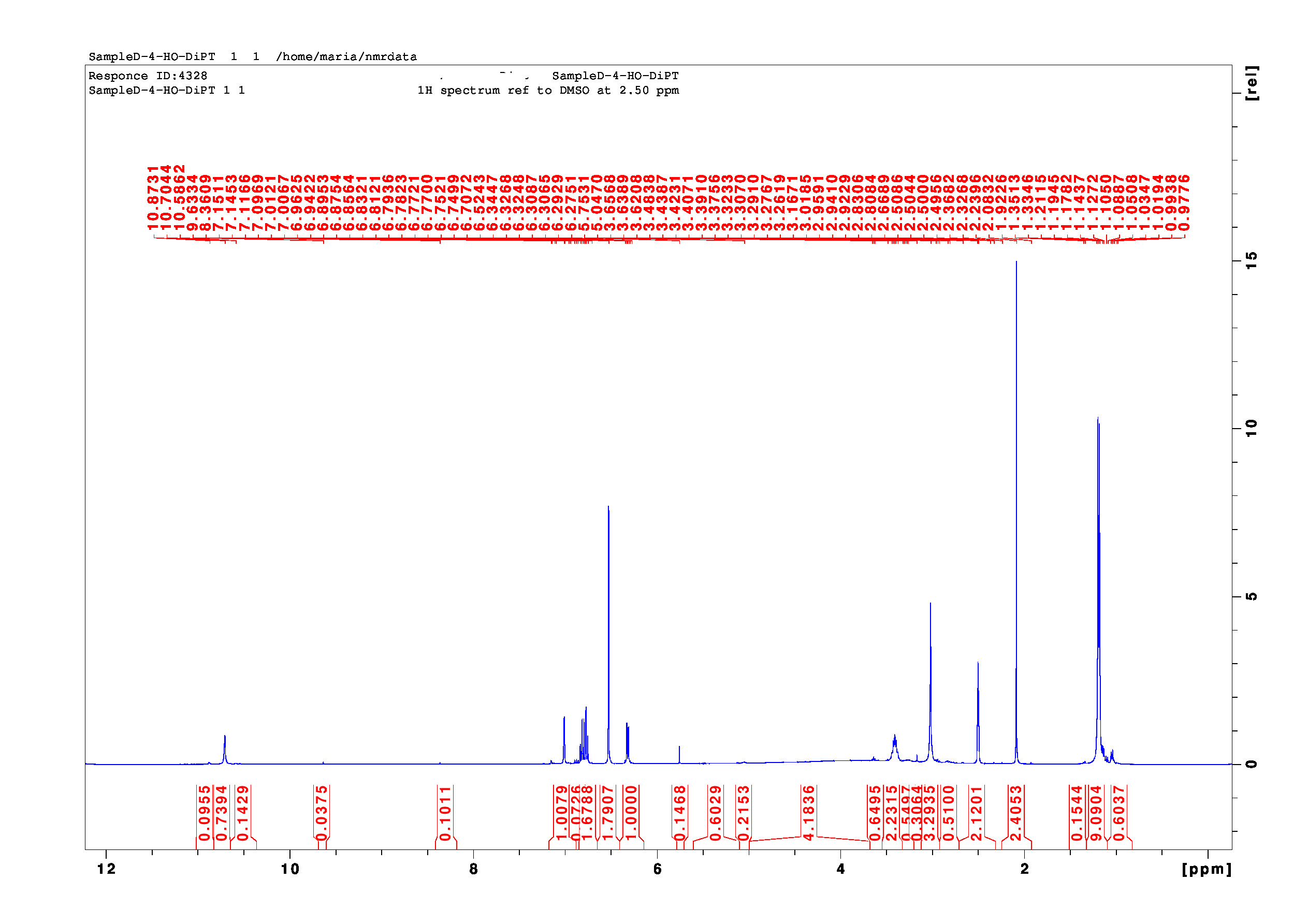
Hi, I might be able to comment on this one. The 1H NMR you provided shows signals that are corresponding to a 4-hydroxyindole-type compound. The post-processing of the 1H NMR spectrum is poor. The compound is fairly clean, but it is not 4-HO-DIPT.
I will try to walk you through my analysis assuming some knowledge in chemistry and none in NMR spectroscopy. The 1H NMR spectrum shows ONLY the hydrogens of the molecule. Key data are 1) “chemical shifts” in [ppm] – the position of a signal on the x-scale indicated by the red numbers above the blue curve of the spectrum, 2) “integrals” – the area under a signal that is proportional to the number of protons that are responsible for a given signal, indicated by the red numbers and designated areas under the blue curve of the spectrum… In addition, 1H NMR spectra provide 3) “multiplicities” – the patterns of each signal that indicate the number of adjacent protons, and 4) “coupling constants” – the distance between individual lines of a signal, which indicate the kind and angle of bonds between the protons. Both, multiplicities and coupling constants require well resolved spectra and are not required for this basic discussion.
HEMIFUMRATE - The integrals of the 1H NMR show a signal at 6.3 ppm for a proton of the indole ring that has been integrated to 1.00 H (by definition) and a proton for the fumarate double bond at 6.5 ppm that integrates to 1.8H. Because fumarate has two identical protons that both give the signal at 6.3 ppm this translates roughly to a 1 / 0.9 mixture of an indole-type compound and fumarate. That is OK, but one fumarate can neutralize two tryptamine molecules, here they only neutralize one – you have a “hemifumarate”. It’s still acidic and often less stable in the long-term. Furthermore, there is more fumarate and less indole base for a given mass of final material. They sell you more fumarate and less base this way. Might be a common reason why some material is less potent than others.
POOR NMR PROCESSING - It is a bad 1H NMR – the area 3.5 – 5.0 ppm integrates to (4.2H) – that would be a 420% impurity… but there is not even a real signal in this area. The operator “maria” did no process these data correctly. Once NMRs are measured the data need to be processed (Fourier-transformed, phase corrected, baseline corrected) before they are analyzed (peak picking, integration). In this case “maria” skipped the baseline correction, which is essential for proper integration results. As a consequence, the noise at 3.5 – 5.0 ppm now integrates to 4.2H. Therefore, this NMR is not suitable for a reliable quantitative assessment. But of course, we can judge if the sample is clean or not – we simply cannot quantify exactly how clean it is.
IMPURITIES - There are a couple of impurities, these are visible as small signals all over the spectrum. To judge their relative amounts, we have to compare the integrals of our target molecule (the tryptamine) and the impurities. All impurities integrate to considerably less than 1H (of course they could integrate to more than 1H in case there were major impurities – here they are minor). For example, there are impurities at 1.0 ppm (0.6H), 3.2 ppm (0.6 H), 5.5 ppm (0.15H), 8.4 ppm (0.1H), 9.7 ppm (0.03H) plus some others. Many of these might be residual solvents. They are minor – I would personally not be too concerned about them.
WRONG COMPOUND - Having said all of this regarding your initial question I would now like to come to the main problem with this NMR… because it establishes unambiguously that the sample is NOT the desired compound 4-HO-DIPT. DIPTs have two N-linked isopropyl-groups…, every isopropyl group has two methyl groups… (that makes 2 * 2 = 4 methyl groups), and every methyl group has three hydrogens… (that makes 2 * 2 * 3 = 12 hydrogens). Consequently, the signal for the methyl groups at 1.2 ppm has to integrate to “12H”. But it only integrates to “9H” in the provided NMR. Furthermore, there is a prominent signal at about 2.1 ppm (2.4H) that totally contradicts the assumed structure. This signal might be corresponding to an N-acetyl group, but it is clearly NOT bound to an oxygen (like in 4-AcO-DIPT). Finally, the remaining signals at 3.0 ppm (2.2H) and 3.4 ppm (3.3H) do not add up for the remaining two methylene groups (2 x 2H) plus the two methine groups (2H) that would be required for a DIPT-like molecule. Without proper post-processing and reliable integrals, it is impossible to say what this compound is instead.
There is an excellent 1H NMR spectrum of 4-HO-DIPT provided by a prominent vendor for these types of molecules that you might want to compare with. But considering that this is my first post I cannot even upload it here and I do not want to start my time on bluelight by getting into trouble for “sourcing”… I leave it like this and hope you will be able to find it yourself.
CONCLUSION – the NMR measured by “maria” contradicts the claim that this material is 4-HO-DIPT. This demonstrates that the availability of analytical data (like NMR spectra) by a vendor means nothing with respect to sample purity or identity because customers (as well as vendors) are usually not qualified to interpret these data.
TL;DR: rip-off material that is excessively rich in fumarate, quite clean, but not the right compound. Botched underground synthesis or totally irresponsible commercial vendor. sorry.