How toxic is ibogaine?
Litjens RP, Brunt TM
Ibogaine and noribogaine interact with multiple neurotransmitter systems, and show micromolar affinity for N-methyl-D-aspartate (NMDA),
K- and u-opioid receptors and sigma-2 receptor sites. Furthermore, ibogaine interacts with the acetylcholine, serotonin and dopamine systems, and it alters the expression of several proteins including substance P, brain-derived neurotrophic factor (BDNF), c-fos and egr-1.
NEUROTOXICITY:
Neurodegeneration was shown in rats, probably mediated by stimulation of the inferior olive, which has excitotoxic effects on Purkinje cells in the cerebellum. Neurotoxic effects of ibogaine may not be directly relevant to its anti-addictive properties, as no signs of neurotoxicity were found following doses lower than 25mg/kg intra-peritoneal in rats. Noribogaine might be less neurotoxic than ibogaine.
CARDIOTOXICITY:
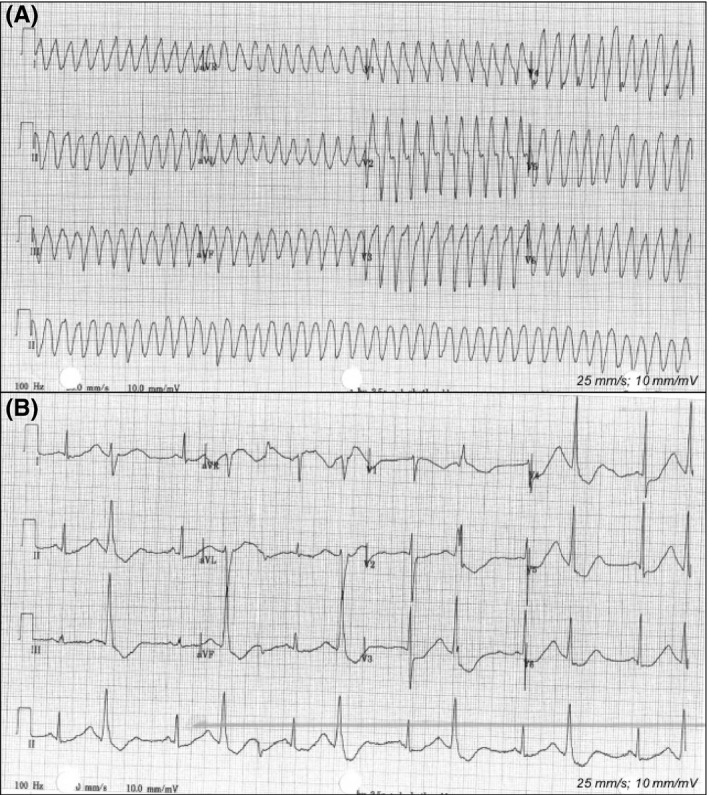
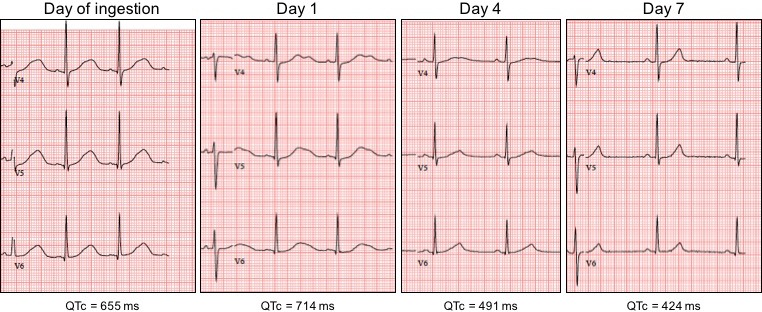
Ether-a-go-go-related gene (hERG) potassium channels in the heart might play a crucial role in ibogaine's cardiotoxicity, as hERG channels are vital in the repolarization phase of cardiac action potentials and blockade by ibogaine delays this repolarization, resulting in QT interval prolongation and, subsequently, in arrhythmias and sudden cardiac arrest. 27 fatalities have been reported following the ingestion of ibogaine, and pre-existing cardiovascular conditions have been implicated in the death of individuals for which post-mortem data were available.
TOXICITY FROM DRUG-DRUG INTERACTION:
Polymorphism in the CYP2D6 enzyme can influence blood concentrations of both ibogaine and its primary metabolite, which may have implications when a patient is taking other medication that is subject to significant CYP2D6 metabolism.
CONCLUSIONS:
The case reports presented here suggest that ibogaine caused ventricular tachyarrhythmias and prolongation of the QT interval in individuals, without any pre-existing cardiovascular condition or family history. Noribogaine appears at least as harmful to cardiac functioning as ibogaine.
Alternative therapists and drug users are still using iboga extract, root scrapings, and ibogaine HCl to treat drug addiction with limited medical supervision. These are risky experiments and more ibogaine-related deaths are likely to occur, particularly in those with pre-existing cardiac conditions and those taking concurrent medications.
https://www.ncbi.nlm.nih.gov/pubmed/26807959
-----
Anti-HERG activity and the risk of drug-induced arrhythmias and sudden death
M.L. De Bruin, M. Pettersson, R.H.B. Meyboom, A.W. Hoes, H.G.M. Leufkens
Drug-induced QTc-prolongation, resulting from inhibition of HERG potassium channels may lead to serious ventricular arrhythmias and sudden death. We studied the quantitative anti-HERG activity of pro-arrhythmic drugs as a risk factor for this outcome in day-to-day practice.
All 284 426 case reports of suspected adverse drug reactions of drugs with known anti-HERG activity received by the International Drug Monitoring Program of the World Health Organization (WHO-UMC) up to the first quarter of 2003, were used to calculate reporting odds ratios (RORs). Cases were defined as reports of cardiac arrest, sudden death, Torsade de Pointes, ventricular fibrillation, and ventricular tachycardia, and compared with non-cases regarding the anti-HERG activity, defined as the effective therapeutic plasma concentration divided by the HERG IC50 value, of suspected drugs. We identified a significant association of 1.93 between the anti-HERG activity of drugs, measured as log10, and reporting of serious ventricular arrhythmias and sudden death to the WHO-UMC database.
Conclusion: Anti-HERG activity is associated with the risk of reports of serious ventricular arrhythmias and sudden death in the WHO-UMC database. These findings are in support of the value of pre-clinical HERG testing to predict pro-arrhythmic effects of medicines.
Introduction
Drug-induced prolongation of the QTc-interval usually results from concentration-dependent blocking of cardiac HERG potassium channels.1,2 An excessively prolonged QTc-interval can, under the right circumstances, lead to a polymorphic ventricular arrhythmia known as Torsade de Pointes (TDP). When TDP is sustained, symptoms arising from impaired cerebral circulation, such as dizziness, syncope, and/or seizures, may become manifest. TDP can subsequently degenerate into ventricular fibrillation and, not uncommonly, cardiac arrest or sudden death may occur. It is not clear what percentage of TDP arrhythmias are non-sustained and what percentage degenerate into ventricular fibrillation. Over the last decade, this adverse reaction has attracted considerable clinical and regulatory interest and has been the most common cause of withdrawal, or restriction of the use, of drugs on the market.
Consider’ document which made recommendations for non-clinical and clinical approaches to assess the risk of QTc-interval prolongation and TDP for non-cardiovascular drugs. The strategies described are now being harmonized by the International Conference of Harmonization (ICH), and a draft version of the ‘Note for Guidance’ document is currently available. Based on these regulatory recommendations, most new drugs are tested nowadays for their ability to block HERG potassium channels and rapid potassium currents (IKr). However, there is still much debate going on within the pharmaceutical industry, as well as regulatory authorities, on the predictive value of HERG channel binding and the risk of cardiac arrhythmias. For example, collaborating researchers from several pharmaceutical industries recently published an extensive overview of 100 QTc-prolonging drugs and their ability to bind to HERG-channels in relation to free-plasma concentrations.
These authors related this anti-HERG activity to the tor-sadogenic propensities of the drugs. Drugs were assigned to one of the following five categories of decreasing tor-sadogenicity: (i) anti-arrhythmic drugs, (ii) drugs withdrawn or suspended due to TDP risk, (iii) drugs with measurable TDP risk in humans or many TDP case reports in published literature, (iv) isolated TDP case reports, and (v) no published reports of TDP in humans, but with a certain degree of suspicion because of, for example, therapeutic class, drug interactions, etc. The clinical relevance of this type of categorization, however, remains to be confirmed.
Previously, several studies have shown that female gender is a rather strong predictor for drug-induced TDP, since 70% of the published case reports concerned women. In the present study, we found that (of the ADR reports with known gender of the patient) 56% of the case patients were female, compared with 58% of the patients experiencing other ADRs. We used, however, a composite endpoint which included, apart from TDP: cardiac arrest, sudden death, ventricular fibrillation, and ventricular tachycardia. When we focused solely on the ADR-reports of TDP, we found that more than 68% of these reports concerned females.
General limitations of the dataset should be discussed. First, the study was restricted to drugs for which HERG binding properties as well as therapeutic free plasma concentrations have been studied, and published. The number of drugs being tested for HERG-activities is still increasing and these analyses should be repeated when more data are available. Secondly, the ETCPunbound/1C50 ratios were based on therapeutic plasma levels at recommended doses. The case reports in the WHO-UMC database, however, do not disclose in sufficient detail the doses used by these patients. Plasma Levels may increase when pharmacokinetic drug–drug interactions occur or alternative routes of administration are used. Specific anti-HERG activities were onLy known for terfenadine plus CYP3A4 inhibitors, and iv erythromycin. Uncertainty of actual plasma levels may have influenced our results.
The method of reaction proportion signalling has several drawbacks. ADRs were reported on a voluntary basis, and therefore represented only a fraction of the actual adverse events that occurred. Selective under- and over-reporting of particular ADRs within the overall under-reporting can Lead to misinterpretations when comparing drugs with respect to ADRs. ADRs which are more likely than others to be reported are ADRs of relatively new drugs severe ADRs, and ADRs which are not listed in the summary of product characteristics. All these aspects can be seen in the subgroup analyses we performed. The association was stronger shortly after marketing, and it was Less well pronounced among patients taking anti-arrhythmic drugs, for which the pro-arrhythmic side-effects have been already described. The association weakens when the study event is Less severe (syncope vs. ventricular arrhythmia). Another factor which may have influenced our results is selective reporting as a result of media attention. This factor has been described previously for the association between cardiac arrhythmias and the use of anti-histamine drugs, and similarly in our study the association between exposure and outcome is stronger after 1 January 1998 than before.
We did not, of course, study the effects of individual drugs, but the in vitro anti-HERG activities of drugs, and combinations of drugs. These molecular properties of drugs are unlikely to be known by healthcare providers in daily practice. We therefore think that we have used a more objective exposure measure, which is less susceptible to recognized bias. Moreover, all sub-group analyses point in a similar direction and negative control outcomes which should not be reLated to anti-HERG activity (hepatitis, skin ulcer) are indeed unrelated to the exposure. We therefore believe that our findings represent a true connection.
There were several drugs that appeared not to follow the predicted association. Their observed cases/non-cases ratios were relatively high or Low compared with the ratios fitted by our logistic model. For ibutilide, bepridil, amiodarone, sotalol, and flecainide, the cases/non-cases ratio is higher than expected. These drugs are prescribed to patients with cardiac diseases and therefore ‘confounding by indication’ may have caused this relatively high fraction of ‘case-events’. In addition, only less than 200 case reports were used to estimate the ratio for ibutilide and bepridil. Slightly more than 300 case reports were used to estimate the cases/non-cases ratio for combination of terfenadine and CYP3A4 inhibiting drugs. This relatively high ratio may have been caused by selective reporting of ADRs due to media attention for cardiac arrhythmias associated with the combined use of these drugs.
For ketoconazole, mefloquine, and aprindine the fraction of ‘case-events’ in the WHO-UMC database is much lower than expected, based on anti-HERG activity. These drugs could be regarded as drugs with ‘false positive’ anti-HERG activities. For ketoconazole this effect was described before. However, the low ratio may also be explained by the fact that there were relatively many ADR reports of ‘skin and appendages disorders’ as well as ‘liver and biliary system disorders’, competing with the ADRs of our interest to stand out is proportionately against all other case reports. Both ADRs counted for 23% of all ADRs that were reported for ketoconazole, whereas the percentages among the case reports of all studied drugs together were 12% and 4%, respectively. Similarly the relatively high proportion of case reports of ‘psychiatric disorders’ (31% for mefloquine vs. 13% overall) and ‘central and peripheral nervous system disorders’ (25% for mefloquine vs. 14% overall) could have competed with the ADRs of our interest.
Drugs that bind to HERG potassium channels in concentrations close to or lower than therapeutic plasma concentrations (i.e. have a high Log10 ETCPunbound/1C50 ratio) have a high risk of reports of serious ventricular arrhythmias, and sudden death, in the WHO-UMC database, indicating a higher pro-arrhythmic risk. The higher the 1C50 (toxic drug level) compared with the ETCP unbound value (therapeutic drug level), the higher this risk. These findings support the vaLue of pre-clinical HERG testing for predicting proarrhythmic effects of medicines.
https://pdfs.semanticscholar.org/dc...8.2125483196.1540870256-1162897731.1540369811
") Just ran across this:
Just ran across this: