JohnBoy2000
Bluelighter
- Joined
- May 11, 2016
- Messages
- 2,463
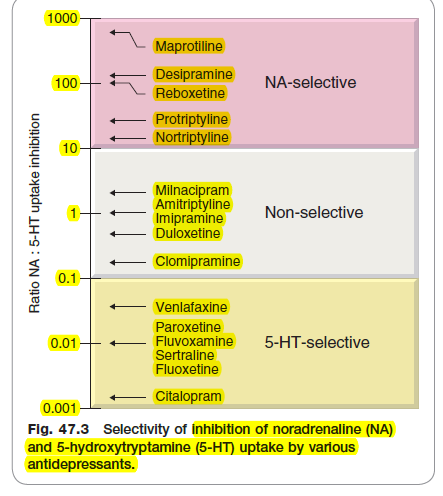
The theoretical determination of drug efficacy - and correct me if I'm wrong here but - it would be, the receptor affinity profile of the drug, in terms of identifying specifically what it binds to - the Ki values for each receptor subtype;
Viewed in perspective of the receptor occupancy relative to each receptor subtype - right?
So by example - reboxetine and atomoxetine are both highly selective, and highly potent, for the noradrenergic transporter.
Their Ki values for this target and not hugely different, though slightly higher affinity lies with atomoxetine.
However, in perspective of receptor occupancy - the clinical description of "highly selective and highly potent" is markedly different for both.
In terms of clinically applicable doses - reboxetine doesn't come close to target saturation. It's receptor occupancy is rather weak.
Atomoxetine - saturates the NA transporter at even quite low doses - but this troughs quickly at low doses; higher doses provide a more sustained occupancy.
However - for older drugs - desipramine by example - I simply can't find any info relate to receptor occupancy.
There are studies that claim it's the most potent noradrenergic agent available - but without any figures to seemingly demonstrate this?
I've been running searches on google scholar and using sci hub to unlock papers.
I did find one study for an old tricyclic, nortriptyline - it's receptor occupancy for NET was like, 40% at full dose.
Yet it's marketed as a potent NA agent.
Kind of misleading - no?
Is there a data base or something that has receptor occupancies for the various drugs?
Viewed in perspective of the receptor occupancy relative to each receptor subtype - right?
So by example - reboxetine and atomoxetine are both highly selective, and highly potent, for the noradrenergic transporter.
Their Ki values for this target and not hugely different, though slightly higher affinity lies with atomoxetine.
However, in perspective of receptor occupancy - the clinical description of "highly selective and highly potent" is markedly different for both.
In terms of clinically applicable doses - reboxetine doesn't come close to target saturation. It's receptor occupancy is rather weak.
Atomoxetine - saturates the NA transporter at even quite low doses - but this troughs quickly at low doses; higher doses provide a more sustained occupancy.
However - for older drugs - desipramine by example - I simply can't find any info relate to receptor occupancy.
There are studies that claim it's the most potent noradrenergic agent available - but without any figures to seemingly demonstrate this?
I've been running searches on google scholar and using sci hub to unlock papers.
I did find one study for an old tricyclic, nortriptyline - it's receptor occupancy for NET was like, 40% at full dose.
Yet it's marketed as a potent NA agent.
Kind of misleading - no?
Is there a data base or something that has receptor occupancies for the various drugs?