Thou
Bluelighter
- Joined
- Mar 10, 2007
- Messages
- 10,860
Well, it looks like they finally finished removing me as a mod. It was fun guys!
That's a shame. Thank you Nuke for all your smarts and wisdom
N&PD Moderators: Skorpio | thegreenhand
Well, it looks like they finally finished removing me as a mod. It was fun guys!
I am mostly interested in organic chemistry and my interest in pharmacology is a result of this and my interest in how our whole world works like. I've recently started studying chemistry again. I had a 3-year break due to various health problems and I didn't manage to get a master's degree. I even started studying IT during the break, but I quit it when I realised chemistry is my biggest passion and I want to learn it because it simply makes me happy. I don't really care about the money I could earn in the future, it's not a factor at all, but I'm definitely not a person who would accept an underpayment. I seriously consider staying at some university but I still have plenty of time and perhaps I will have a better opportunity to research. As I was thinking about it a few days ago, it made me kind of scared when I realised I actually hadn't planned studying chemistry again. It would be such a shame because studying IT didn't make me happy at all, it was such a tiresome duty. I'm really glad I've started changing my life so I can live the way I've always wanted. The past doesn't matter but the knowledge got through past experiences.
Why are you nervous to reveal what you study? :>
Stimulants don't effect people with ADD any differently than others
https://www.nature.com/articles/1301164
Stimulants such as methylphenidate and amphetamine are currently the most common treatment for attention deficit hyperactivity disorder (ADHD). For years, it was assumed that stimulants had paradoxical calming effects in ADHD patients, whereas stimulating ?€˜normal?€™ individuals and producing locomotor activation in rats. It is now known that low doses of stimulants focus attention and improve executive function in both normal and ADHD subjects. Furthermore, the seminal work of Kuczenski and Segal showed that low, oral doses of methylphenidate reduce locomotor activity in rats as well.
http://www.thelancet.com/journals/lancet/article/PIIS0140-6736(17)32802-7/fulltextWe identified 28 552 citations and of these included 522 trials comprising 116 477 participants. In terms of efficacy, all antidepressants were more effective than placebo, with ORs ranging between 2.13 (95% credible interval [CrI] 1.89-2.41) for amitriptyline and 1.37 (1.16-1.63) for reboxetine. For acceptability, only agomelatine (OR 0.84, 95% CrI 0.72-0.97) and fluoxetine (0.88, 0.80-0.96) were associated with fewer dropouts than placebo, whereas clomipramine was worse than placebo (1.30, 1.01-1.68). When all trials were considered, differences in ORs between antidepressants ranged from 1.15 to 1.55 for efficacy and from 0.64 to 0.83 for acceptability, with wide CrIs on most of the comparative analyses. In head-to-head studies, agomelatine, amitriptyline, escitalopram, mirtazapine, paroxetine, venlafaxine, and vortioxetine were more effective than other antidepressants (range of ORs 1.19-1.96), whereas fluoxetine, fluvoxamine, reboxetine, and trazodone were the least efficacious drugs (0.51-0.84). For acceptability, agomelatine, citalopram, escitalopram, fluoxetine, sertraline, and vortioxetine were more tolerable than other antidepressants (range of ORs 0.43-0.77), whereas amitriptyline, clomipramine, duloxetine, fluvoxamine, reboxetine, trazodone, and venlafaxine had the highest dropout rates (1.30-2.32). 46 (9%) of 522 trials were rated as high risk of bias, 380 (73%) trials as moderate, and 96 (18%) as low; and the certainty of evidence was moderate to very low.
All antidepressants were more efficacious than placebo in adults with major depressive disorder. Smaller differences between active drugs were found when placebo-controlled trials were included in the analysis, whereas there was more variability in efficacy and acceptability in head-to-head trials. These results should serve evidence-based practice and inform patients, physicians, guideline developers, and policy makers on the relative merits of the different antidepressants.
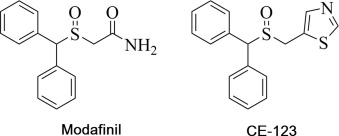
Dopamine reuptake inhibitors have been shown to improve cognitive parameters in various tasks and animal models. We recently reported a series of modafinil analogues, of which the most promising, 5-((benzhydrylsulfinyl)methyl) thiazole (CE-123), was selected for further development. The present study aims to characterize pharmacological properties of CE-123 and to investigate the potential to enhance memory performance in a rat model. In vitro transporter assays were performed in cells expressing human transporters. CE-123 blocked uptake of [3H] dopamine (IC50 = 4.606 uM) while effects on serotonin (SERT) and the norepinephrine transporter (NET) were negligible. Blood-brain barrier and pharmacokinetic studies showed that the compound reached the brain and lower elimination than R-modafinil. The Pro-cognitive effect was evaluated in a spatial hole-board task in male Sprague-Dawley rats and CE-123 enhances memory acquisition and memory retrieval, represented by significantly increased reference memory indices and shortened latency. Since DAT blockers can be considered as indirect dopamine receptor agonists, western blotting was used to quantify protein levels of dopamine receptors D1R, D2R and D5R and DAT in the synaptosomal fraction of hippocampal subregions CA1, CA3 and dentate gyrus (DG). CE-123 administration in rats increased total DAT levels and D1R protein levels were significantly increased in CA1 and CA3 in treated/trained groups. The increase of D5R was observed in DG only. Dopamine receptors, particularly D1R, seem to play a role in mediating CE-123-induced memory enhancement. Dopamine reuptake inhibition by CE-123 may represent a novel and improved stimulant therapeutic for impairments of cognitive functions.
The use of opioids for the treatment of pain, while largely effective, is limited by detrimental side effects including analgesic tolerance, physical dependence, and euphoria, which may lead to opioid abuse. Studies have shown that compounds with a mu-opioid receptor (MOR) agonist/delta-opioid receptor (DOR) antagonist profile reduce or eliminate some of these side effects including the development of tolerance and dependence. Herein we report the synthesis and pharmacological evaluation of a series of tetrahydroquinoline-based peptidomimetics with substitutions at the C-8 position. Relative to our lead peptidomimetic with no C-8 substitution, this series affords an increase in DOR affinity and provides greater balance in MOR and DOR binding affinities. Moreover, compounds with carbonyl moieties at C-8 display the desired MOR agonist/DOR antagonist profile whereas alkyl substitutions elicit modest DOR agonism. Several compounds in this series produce a robust antinociceptive effect in vivo and show antinociceptive activity for greater than 2 h after intraperitoneal administration in mice.


Cebranopadol is a novel potent analgesic agonist at the nociceptin/orphanin FQ peptide (NOP) and classical opioid receptors. As NOP receptor activation has been shown to reduce side effects related to the activation of μ‐opioid peptide (MOP) receptors, the present study evaluated opioid‐type physical dependence produced by cebranopadol in mice and rats. In a naloxone‐precipitated withdrawal assay in mice, a regimen of seven escalating doses of cebranopadol over 2 days produced only very limited physical dependence as evidenced by very little withdrawal symptoms (jumping) even at cebranopadol doses clearly exceeding the analgesic dose range. In contrast, mice showed clear withdrawal symptoms when treated with morphine within the analgesic dose range. In the rat, spontaneous withdrawal (by cessation of drug treatment; in terms of weight loss and behavioral score) was studied after 4‐week subacute administration. Naloxone‐precipitated withdrawal (in terms of weight loss and behavioral score) was studied in the same groups of rats after 1‐week re‐administration following the spontaneous withdrawal period. In both tests, cebranopadol‐treated rats showed only few signs of withdrawal, while withdrawal effects in rats treated with morphine were clearly evident. These findings demonstrate a low potential of cebranopadol to produce opioid‐type physical dependence in rodents. The prospect of this promising finding into the clinical setting remains to be established.
Long-term use of potent cannabis during adolescence increases the risk of developing schizophrenia later in life, but to date, the mechanisms involved remain unknown. Several findings suggest that the functional selectivity of serotonin 2A receptor (5-HT2AR) through inhibitory G-proteins is involved in the molecular mechanisms responsible for psychotic symptoms. Moreover, this receptor is dysregulated in the frontal cortex of schizophrenia patients. In this context, studies involving cannabis exposure and 5-HT2AR are scarce. Here, we tested in mice the effect of an early chronic Δ9-tetrahydrocannabinol (THC) exposure on cortical 5-HT2AR expression, as well as on its in vivo and in vitro functionality. Long-term exposure to THC induced a pro-hallucinogenic molecular conformation of the 5-HT2AR and exacerbated schizophrenia-like responses, such as prepulse inhibition disruption. Supersensitive coupling of 5-HT2AR toward inhibitory Gαi1-, Gαi3-, Gαo-, and Gαz-proteins after chronic THC exposure was observed, without changes in the canonical Gαq/11-protein pathway. In addition, we found that inhibition of Akt/mTOR pathway by rapamycin blocks the changes in 5-HT2AR signaling pattern and the supersensitivity to schizophrenia-like effects induced by chronic THC. The present study provides the first evidence of a mechanistic explanation for the relationship between chronic cannabis exposure in early life and increased risk of developing psychosis-like behaviors in adulthood.
Objective: The study evaluates nutmeg fractions for binding capacity with various CNS receptors and their potential interaction with the endocannabinoid system.
Materials and methods: Dichloromethane (DF) and ethyl acetate (EF) fractions were prepared from the methanol extract of powdered whole nutmeg. The HPLC-profiled fractions were assayed by the NIMH Psychoactive Drug Screening Program (PDSP) in a panel of CNS targets at a 10 μg/mL concentration. The fractions were also screened for fatty acid amide hydrolase (FAAH) and monoacylglycerol lipase (MAGL) inhibition, initially at a concentration of 500 μg/mL, then by concentration-dependent inhibition studies.
Results: None of the tested fractions showed significant binding to CNS receptors included in the PDSP panel. However, both fractions exerted significant inhibition of the FAAH and MAGL enzymes. The DF fraction inhibited FAAH and MAGL enzymes at IC50 values of 21.06 ? 3.16 and 15.34 ? 1.61 μg/mL, respectively. Similarly, the EF fraction demonstrated FAAH and MAGL inhibition with IC50 values of 15.42 ? 3.09 and 11.37 ? 6.15 μg/mL, respectively.
Methamphetamine (METH) is a drug with a high addictive potential that is widely abused across the world. Although it is known that METH dysregulates both dopamine transmission and dopamine reuptake, the specific mechanism of action remains obscure. One promising target of METH is the sigma receptor, a chaperone protein located on the membrane of the endoplasmic reticulum. Using fast-scan cyclic voltammetry, we show that METH-enhancement of evoked dopamine release and basal efflux is dependent on sigma receptor activation. METH-induced activation of sigma receptors results in oxidation of a cysteine residue on VMAT2, which decreases transporter function. Unilateral injections of the sigma receptor antagonist BD-1063 prior to METH administration increased dopamine-related ipsilateral circling behavior, indicating the involvement of sigma receptors. These findings suggest that interactions between METH and the sigma receptor lead to oxidative species (most likely superoxide) that in turn oxidize VMAT2. Altogether, these findings show that the sigma receptor has a key role in METH dysregulation of dopamine release and dopamine-related behaviors.
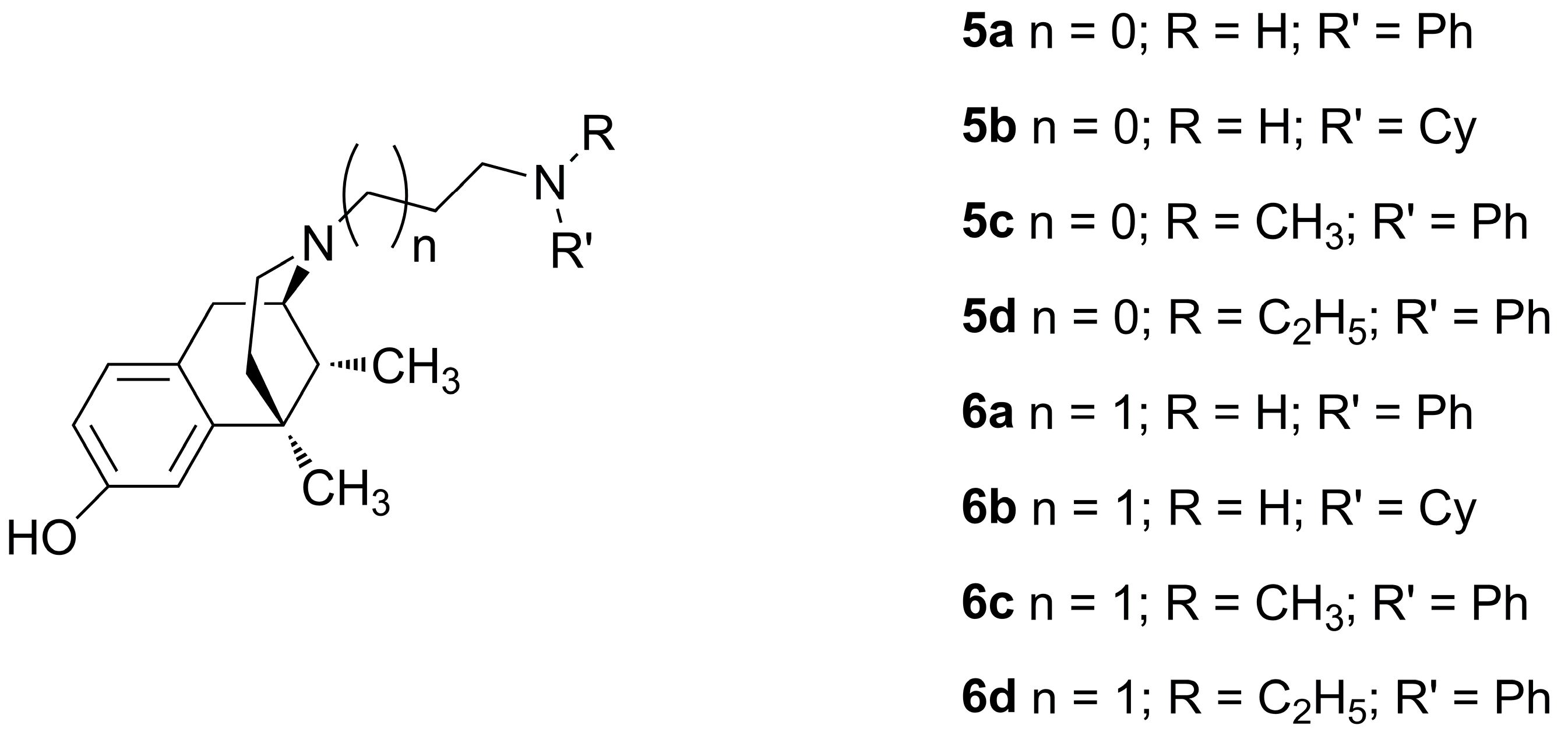
The opioid pharmacological profile of cis-(−)-N-normetazocine derivatives is deeply affected by the nature of their N-substituents. Here, our efforts were focused on the synthesis and pharmacological evaluation of novel derivatives of the lead LP1, a multitarget opioid analgesic compound featuring an N-phenylpropanamido substituent. LP1 derivatives 5a–d and 6a–d were characterized by flexible groups at the N-substituent that allow them to reposition themselves relative to cis-(−)-N-normetazocine nucleus, thus producing different pharmacological profiles at the mu, delta and kappa opioid receptors (MOR, DOR and KOR) in in vitro and in vivo assays. Among the series, compound 5c, with the best in vitro and in vivo profile, resulted a MOR agonist which displays a KiMOR of 6.1 nM in a competitive binding assay, and an IC50 value of 11.5 nM and an Imax of 72% in measurement of cAMP accumulation in HEK293 cells stably expressing MOR, with a slight lower efficacy than LP1. Moreover, in a mouse model of acute thermal nociception, compound 5c, intraperitoneally administered, exhibits naloxone-reversed antinociceptive properties with an ED50 of 4.33 mg/kg. These results expand our understanding of the importance of N-substituent structural variations in the opioid receptor profile of cis-(−)-N-normetazocine derivatives and identify a new MOR agonist useful for the development of novel opioid analgesics for pain treatment.