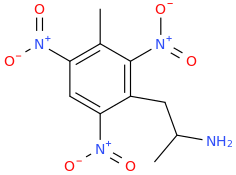
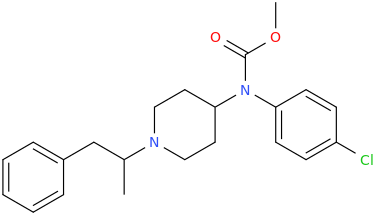
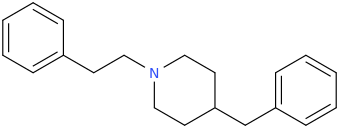
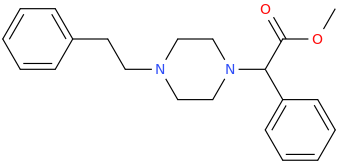
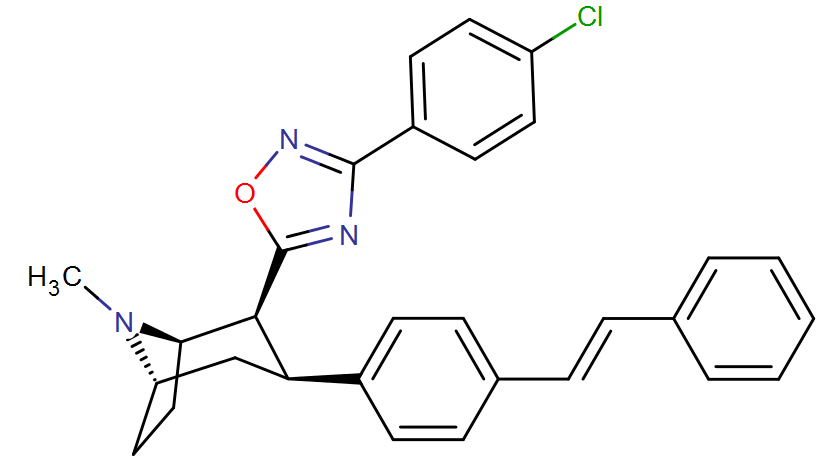
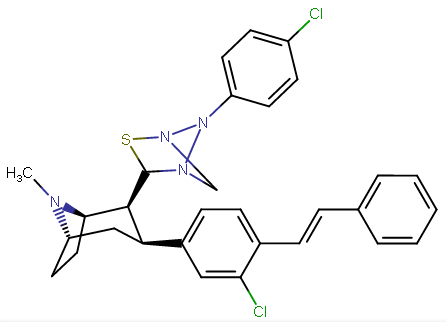
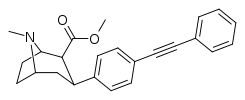
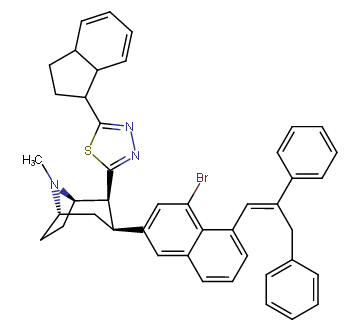
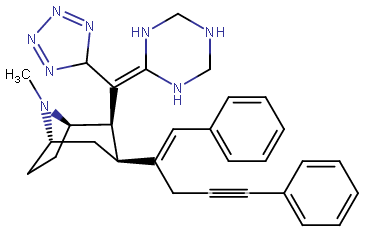
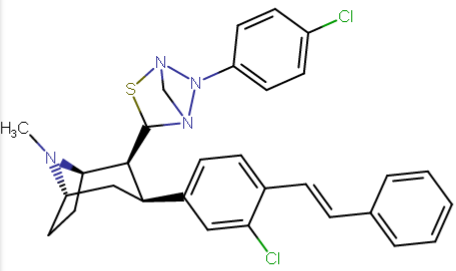
RTI-143 (NET/DAT selectivity ratio of 9,919,
over nine-thouuuuusand!) @ 2-beta substitution + RTI-436 (634.6 NET/DAT ratio) @ 3-beta para-benzene position.
Furthermore, if you
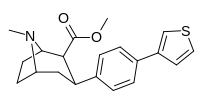
compare RTI-195 to RTI-199 (pg. 16, bottom two), where the only difference is a sulfur on the latter to the oxygen on the shared heterocycle at the C2 pos., it changes NET value from 1,310 to 24,320.8 (however, the substitution pattern in the heterocycle is different than in RTI-143, and since electrostatic factors contribute in the case of C2, I'm adding rather than omitting, but without making a heterocylic di-aryl (which one would think to have had an example somewhere in literature that someone studying this nigh a decade as I have would have come across but have not so figure it'd gimp the structure completely) and keeping the electrostatic sub-pattern the same have added an internal-to-cyclopentane diaziridine to the nitrogens; meaning also putting in a near chlorine sub. on the aryl like in RTI-143 also as the di-subs show little difference between the para and meta when it comes to halogens with PTs @ the C3, so I added the C3 chlorine without forgoing the 436's para-(trans)stilbene skeleton. So taking that into account:
(though looking at its least energy state, it's more of some trilactam than a diaziridine... trilactam, sounds very "Revenge Of The Nerds-ish")
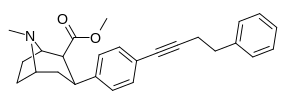
Also an extreme extended length para-pos., rigid triple bond terminating in a second phenyl on the PT C3 phenyl (namesake 'phenyl' phenyl with a side-chain phenyl attached thereon) has much higher binding than expected when longer substitutions have otherwise impaired binding from there, making it be
postulated that there is another remote binding domain on DAT far off from the 180 degree nominally usual ending of the inherent PT phenyl's binding range
i.e.
third best = 3.7 @ DAT binding⤴
2nd Best = 3.09 @ DAT binding⤴
BEST = 1.82 @ DAT binding⤴
fourth best, or "worst of the triple-phenyl-terminating" = 6.28 @ DAT binding (still good! compared to..)⤴
Whereas for example:
tamagnan is half as potent as the worst above w/ a value of 12 @ DAT⤴
and
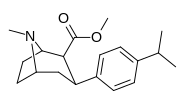
isopropyl-para-PT is a shockingly low 597 (five-hundred-ninety-seven) @ DAT ligand affinity(!)⤴
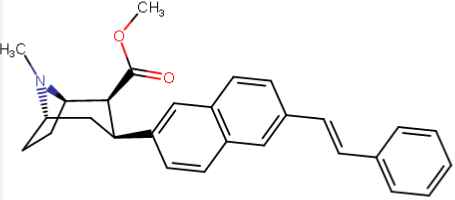
So following this rationale, is there anybody who has access to a 3D rendering program who could tell which of the triple-phenyl-ending-para substitutions (specifically the best / second longest one third down from top) best overlays with a 2-naphthyl substituted PT (or "NT" as it were, i.e. naphthyltropane, just perhaps?) with what would be a similar, equivalent (in 3D) addition but instead on its 6, 7 or 8 position (since it's a naphthyl there's a lot more than the phenyl, of course)?
For instance:
...could someone come up with something potentially better than the above using the naphthyl idea? (since the PT naphthyl alone, without making use of the putative "extra" binding domain on DAT off along in space beyond the phenyl, has a DAT affinity of 0.51, a SERT of 0.80, and a NET of 21.1, such might be very promising if such another binding domain is being used)
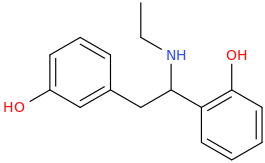
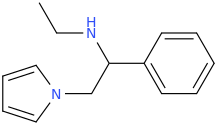
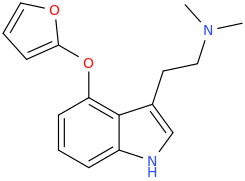



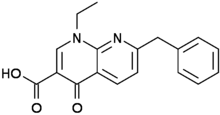
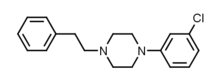
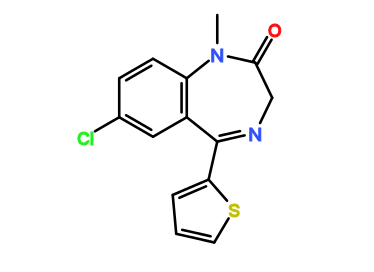
")