fastandbulbous
Bluelight Crew
- Joined
- Jul 29, 2004
- Messages
- 21,304
Factors that influence the structure-activity relationship (SAR) of psychedelic drugs with respect to their agonist activity and binding at the 5HT2A receptor site - a possible new series of high potency 5HT2A agonists
The 5HT2A receptor is possibly the most important when it comes to investigating the actions of psychedelic drugs. These drugs belong almost exclusively to the phenethylamine, tryptamine and ergoline groups of compounds. The other sites that are believed to be important in how they exert their activity are the 5HT1A and 5HT2C receptors (the arrangement of the sites important for binding of compounds to the 5HT2C site are remarkably similar to those of the 5HT2A receptor).
In binding studies using cloned 5HT2A receptor, the drug(s) used in assays using radiolabelled markers are DOB and DOI (4-bromo-2,5-dimethoxyamphetamine and 4-iodo-2,5-dimethoxyamphetamine, respectively). SAR studies carried out by A Shulgin, and documented in the book PIHKAL have shown the following to be needed for maximum activity:
A ring substitution pattern that has methoxy groups at positions 2 and 5 on the benzene ring. Any variation, by either substitution of ethoxy groups for one or both methoxy groups, or substitution into the benzene ring in a pattern other than at position 2 and 5, leads to a reduction of activity; in most cases, this produces a marked fall in activity.
Substitution into the benzene ring at the 4 position with a hydrophobic group. Compounds formed by the use of any of the following groups have shown activity at doses under 100mg in man: alkoxy; alkyl; halogeno or thioalkyl. In order of the potency of the compound formed, they show the following pattern
Most potent… halogeno > alkyl > thioalkyl > alkoxy …least potent.
Within those groups, the more hydrophobic the group, the higher the activity. This is only limited by stearic considerations when the groups become very large. So for each group, the ordering is as follows:
Halogens Iodo > bromo > chloro >> fluoro
Alkyl Propyl > ethyl > methyl >> butyl >> pentyl(amyl)
Thioalkyl Propylthio > ethylthio > methylthio
With the alkoxy substituents, the nature of the intoxication seems to change qualitatively as the group gets bigger.
On the side chain, bearing the ethylamine function, an alpha methyl group (amphetamines) gives greatest activity, followed by the unsubstituted chain (phenethylamines). Substitution into the alpha position by a group larger than methyl abolishes any psychedelic activity.
Alpha substitution activity… methyl > hydrogen >>> ethyl.
Using the above data from PIHKAL, the most potent compound is either DOI or DOB, as both were fully active at about 2.5mg. Fig 1 shows how the relevant substitution pattern interacts with the 5HT2A receptor (diagram modified from original present in a paper by D Nichols)

Further developing the SAR theme started by Shulgin, Nichols produced some conformationally restricted analogues of DOB (they’ve been nicknamed the “dragonfly” compounds, most probably because of their structural resemblance to said winged insects). Fig 2 shows the structure of a couple of the “dragonfly” compounds, and DOB for comparison. Also given are receptor binding data for the three compounds.

As can be seen from the binding data, both of the dragonflies are more active than DOB, the three ringed, fully aromatic compound being the most potent (the dihydrofuran derivative is fully active in man at a dosage of 800-1000ug (0.8-1.0mg), and the fully aromatic furan derivative should be active at an even smaller dosage in man). The lone pairs of electrons on the oxygen atoms of the methoxy groups have been prevented from rotation by incorporation into a furan ring system fused with the benzene ring. The fully aromatic compound shows even more activity, and this is thought to be because all of the atoms of the three rings lie on the same plane. This is thought to be needed for maximum activity (with tryptamines, the indolic nitrogen is in the same space as the oxygen of the 5-methoxy group. Only difference is that nitrogen has only 1 lone pair electrons. The 2-methoxy group corresponds to the 5-hydroxy group of serotonin)
Fig 3 shows how the fully aromatic dragonfly compound would interact with the 5HT2A receptor, and its structure is compared with LSD (the aromatic dragonfly compound’s structure has been overlaid that of LSD for easier comparison. Also, the structure of 5HT/serotonin has also been overlaid that of LSD).

It can be speculated that one of the reasons that LSD shows such high potency as a psychedelic agent in man is because all of the atoms of the indole nucleus, and that of the ring directly attached to it (rings a, b and c – see fig 5), all lie in the same plain, hence presenting a flat face to the receptor. Reduction of the double bond of the other ring (the d ring, that contains the tertiary amine function), abolishes the activity of LSD. Reduction of this bond also removes the conformational restraint that holds the a, b and c ring in the same plane, and the carbon atom at position 4 in LSD is forced either above or below the plane of the indole nucleus. This pushes it into the region of stearic occlusion, which in turn abolishes any activity. It is this area of stearic occlusion that is responsible for the loss of activity when an alpha methyl group is replaced by an alpha ethyl group in the phenethylamine hallucinogens (and why the 2-aminotetralin derivatives are devoid of activity – the saturated ring would not be flat, but would take a modified version of the boat/chair form that cyclohexane exhibits)..
Fig 4 shows a comparison of activity of the 2,5-dimethoxy-4-methylamphetamine (DOM) that occurs with different substitution patterns into the sidechain ethylamine function. As expected, there is a loss of activity that comes with the loss of the alpha-methyl group to give the compound 2C-D. Movement of the methyl group from the alpha position to the beta position causes activity to drop off dramatically (beta-methyl 2C-D). Replacement of the beta-methyl group with a beta-methoxy group (to give BOD), shows a twofold increase in activity over 2C-D. This being the case, the lack of activity of beta-methyl 2C-D cannot be explained as being due to stearic hindrance, as the methoxy group is quite a bit larger than the methyl group. Other than the size of the groups, the other main difference is that the oxygen atom of the methoxy group has two lone pairs of electrons, whereas the methyl group has none.

At this point, it is advantageous to look at the comparison of two tryptamine hallucinogens, in order to shed some light on the activities of the DOM/2C-D derivatives. Comparing N,N-dimethyltryptamine (DMT), with its 4-hydroxy derivative (psilocin), one would expect to see a reduction in activity in psilocin, as the polar OH group would reduce the ability to cross the blood brain barrier: What in fact is seen is a 5 fold increase in activity.

Fig 5 shows the structural configuration of psilocin and BOD superimposed over that of LSD. The thing that they have in common is that the oxygen atom in each compound is in a position to donate lone pairs of electrons into the space that would be occupied by the delocalised pi electrons of the double bond in LSD. The lack of activity of beta-methyl 2C-D confirms that there needs to be negative charge in the area of the double bond. It comes from the delocalised pi electrons in the case of LSD, and lone pairs in the case of psilocin and BOD. Because beta-methyl 2C-D “sticks an atom” into that space, but without a concentration of negative charge to interact with the positive charge in the receptor protein, it binds much more weakly than either 2C-D (doesn’t try to push a methyl group in there) or BOD (oxygen atom puts negative charge – lone pair electrons – into that space) Fig 5a shows distribution of negative charge (pi electrons) around double bond in d ring.

As well as contributing the negative charge, the double bond holds all 4 carbon atoms in the same plane as the atoms of the benzene ring, which as mentioned earlier is very important for receptor binding. The proposed phenethylamine/amphetamine 5HT2A agonists in fig 6 have the whole phenethyl skeleton held in the same plane due to the conjugation of the pi electrons with the delocalised electrons of the benzene ring, as well as presenting the pi electrons in the correct position. They should be more potent than their corresponding amphetamine derivative, as they will bond more strongly to the receptor. This also removes any complications that might occur with any adrenergic receptor interaction (the beta-hydroxy 2C-D derivative, BOHD caused a large drop in blood pressure – in fact the drug methoxamine has the 2,5-dimethoxy configuration, and a beta-hydroxy group, and it is a potent pressor agent), as the beta-hydroxy oxygen atom is in the same position as the benzylic hydroxy group oxygen atom. The corresponding DOM derivative would be 3-amino-2-(2,5-dimethoxy-4-methylphenyl)butene, and the 2C-D derivative would be 3-amino-2-(2,5-dimethoxy-4-methylphenyl)propene.

Also shown in fig 6 is a modification that retains the double bond, substituted into a tryptamine, but retaining the conjugated system of the aromatic nucleus (indole instead of benzene). This would produce a series of 4-vinyl-N, N-dialkyltryptamines, which again should be more potent than their corresponding 4-hydroxy-N,N-dialkyltryptamine, and as the group is far less polar, so should allow better penetration of the blood brain barrier

Fig 6a shows all these modifications taken to their logical conclusion in the double bond version of the fully aromatic dragonfly molecule. If in this molecule, there is an increase in potency of the same magnitude as would be expected when applied to DOB etc, then it could possibly be a phenethylamine derivative that is on a par with LSD in terms of binding, dose etc
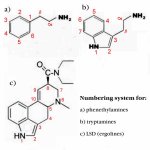
Numbering system of phenethylamines, tryptamines and ergolines is outlined in the jpeg attachment
If anybody can be bothered to plough though all of that, please point if I’ve not seen some obvious flaw in my reasoning
Papers referenced
James J. Chambers, Deborah M. Kurrasch-Orbaugh, Matthew A. Parker, and David E. Nichols. Enantiospecific Synthesis and Pharmacological Evaluation of a Series of
Super-Potent, Conformationally Restricted 5-HT2A/2C Receptor Agonists. J. Med. Chem. 2001, 44, 1003-1010
Nicholas M. Barnes; Trevor Sharp. A review of central 5-HT receptors and their function. Neuropharmacology 38 (1999) 1083–1152
David E. Nichols, Ph.D. The Medicinal Chemistry of Phenethylamine Psychedelics. The Heffter Review of Psychedelic Research, Volume 1, 1998
F. A. B. Aldous, B. C. Barrass, K. Brewster, D. A. Buxton, D. M. Green, R. M. Pinder, P. Rich, M. Skeels, and K. J. Tutt. Structure-Activity Relationships in Psychotomimetic Phenylalkylamines. J.Med. Chem., 2974, Vol. 27, No. 20 1100 - 1111
Nichols D.E, LSD and Its Lysergamide Cousins. The Heffter Review of Psychedelic Research, Volume 2, 2001, 80-87
Shulgin Ann & Alexander T. PIHKAL (Phenethyamines I Have Known And Loved)
-----------------------------------------------------------------------------------------
The final bit was just theoretical pondering on what should happen, but it’ll really need somebody to synthesize it and carry out trials with it to definitely confirm or deny the activity
The 5HT2A receptor is possibly the most important when it comes to investigating the actions of psychedelic drugs. These drugs belong almost exclusively to the phenethylamine, tryptamine and ergoline groups of compounds. The other sites that are believed to be important in how they exert their activity are the 5HT1A and 5HT2C receptors (the arrangement of the sites important for binding of compounds to the 5HT2C site are remarkably similar to those of the 5HT2A receptor).
In binding studies using cloned 5HT2A receptor, the drug(s) used in assays using radiolabelled markers are DOB and DOI (4-bromo-2,5-dimethoxyamphetamine and 4-iodo-2,5-dimethoxyamphetamine, respectively). SAR studies carried out by A Shulgin, and documented in the book PIHKAL have shown the following to be needed for maximum activity:
A ring substitution pattern that has methoxy groups at positions 2 and 5 on the benzene ring. Any variation, by either substitution of ethoxy groups for one or both methoxy groups, or substitution into the benzene ring in a pattern other than at position 2 and 5, leads to a reduction of activity; in most cases, this produces a marked fall in activity.
Substitution into the benzene ring at the 4 position with a hydrophobic group. Compounds formed by the use of any of the following groups have shown activity at doses under 100mg in man: alkoxy; alkyl; halogeno or thioalkyl. In order of the potency of the compound formed, they show the following pattern
Most potent… halogeno > alkyl > thioalkyl > alkoxy …least potent.
Within those groups, the more hydrophobic the group, the higher the activity. This is only limited by stearic considerations when the groups become very large. So for each group, the ordering is as follows:
Halogens Iodo > bromo > chloro >> fluoro
Alkyl Propyl > ethyl > methyl >> butyl >> pentyl(amyl)
Thioalkyl Propylthio > ethylthio > methylthio
With the alkoxy substituents, the nature of the intoxication seems to change qualitatively as the group gets bigger.
On the side chain, bearing the ethylamine function, an alpha methyl group (amphetamines) gives greatest activity, followed by the unsubstituted chain (phenethylamines). Substitution into the alpha position by a group larger than methyl abolishes any psychedelic activity.
Alpha substitution activity… methyl > hydrogen >>> ethyl.
Using the above data from PIHKAL, the most potent compound is either DOI or DOB, as both were fully active at about 2.5mg. Fig 1 shows how the relevant substitution pattern interacts with the 5HT2A receptor (diagram modified from original present in a paper by D Nichols)
Further developing the SAR theme started by Shulgin, Nichols produced some conformationally restricted analogues of DOB (they’ve been nicknamed the “dragonfly” compounds, most probably because of their structural resemblance to said winged insects). Fig 2 shows the structure of a couple of the “dragonfly” compounds, and DOB for comparison. Also given are receptor binding data for the three compounds.
As can be seen from the binding data, both of the dragonflies are more active than DOB, the three ringed, fully aromatic compound being the most potent (the dihydrofuran derivative is fully active in man at a dosage of 800-1000ug (0.8-1.0mg), and the fully aromatic furan derivative should be active at an even smaller dosage in man). The lone pairs of electrons on the oxygen atoms of the methoxy groups have been prevented from rotation by incorporation into a furan ring system fused with the benzene ring. The fully aromatic compound shows even more activity, and this is thought to be because all of the atoms of the three rings lie on the same plane. This is thought to be needed for maximum activity (with tryptamines, the indolic nitrogen is in the same space as the oxygen of the 5-methoxy group. Only difference is that nitrogen has only 1 lone pair electrons. The 2-methoxy group corresponds to the 5-hydroxy group of serotonin)
Fig 3 shows how the fully aromatic dragonfly compound would interact with the 5HT2A receptor, and its structure is compared with LSD (the aromatic dragonfly compound’s structure has been overlaid that of LSD for easier comparison. Also, the structure of 5HT/serotonin has also been overlaid that of LSD).
It can be speculated that one of the reasons that LSD shows such high potency as a psychedelic agent in man is because all of the atoms of the indole nucleus, and that of the ring directly attached to it (rings a, b and c – see fig 5), all lie in the same plain, hence presenting a flat face to the receptor. Reduction of the double bond of the other ring (the d ring, that contains the tertiary amine function), abolishes the activity of LSD. Reduction of this bond also removes the conformational restraint that holds the a, b and c ring in the same plane, and the carbon atom at position 4 in LSD is forced either above or below the plane of the indole nucleus. This pushes it into the region of stearic occlusion, which in turn abolishes any activity. It is this area of stearic occlusion that is responsible for the loss of activity when an alpha methyl group is replaced by an alpha ethyl group in the phenethylamine hallucinogens (and why the 2-aminotetralin derivatives are devoid of activity – the saturated ring would not be flat, but would take a modified version of the boat/chair form that cyclohexane exhibits)..
Fig 4 shows a comparison of activity of the 2,5-dimethoxy-4-methylamphetamine (DOM) that occurs with different substitution patterns into the sidechain ethylamine function. As expected, there is a loss of activity that comes with the loss of the alpha-methyl group to give the compound 2C-D. Movement of the methyl group from the alpha position to the beta position causes activity to drop off dramatically (beta-methyl 2C-D). Replacement of the beta-methyl group with a beta-methoxy group (to give BOD), shows a twofold increase in activity over 2C-D. This being the case, the lack of activity of beta-methyl 2C-D cannot be explained as being due to stearic hindrance, as the methoxy group is quite a bit larger than the methyl group. Other than the size of the groups, the other main difference is that the oxygen atom of the methoxy group has two lone pairs of electrons, whereas the methyl group has none.
At this point, it is advantageous to look at the comparison of two tryptamine hallucinogens, in order to shed some light on the activities of the DOM/2C-D derivatives. Comparing N,N-dimethyltryptamine (DMT), with its 4-hydroxy derivative (psilocin), one would expect to see a reduction in activity in psilocin, as the polar OH group would reduce the ability to cross the blood brain barrier: What in fact is seen is a 5 fold increase in activity.
Fig 5 shows the structural configuration of psilocin and BOD superimposed over that of LSD. The thing that they have in common is that the oxygen atom in each compound is in a position to donate lone pairs of electrons into the space that would be occupied by the delocalised pi electrons of the double bond in LSD. The lack of activity of beta-methyl 2C-D confirms that there needs to be negative charge in the area of the double bond. It comes from the delocalised pi electrons in the case of LSD, and lone pairs in the case of psilocin and BOD. Because beta-methyl 2C-D “sticks an atom” into that space, but without a concentration of negative charge to interact with the positive charge in the receptor protein, it binds much more weakly than either 2C-D (doesn’t try to push a methyl group in there) or BOD (oxygen atom puts negative charge – lone pair electrons – into that space) Fig 5a shows distribution of negative charge (pi electrons) around double bond in d ring.
As well as contributing the negative charge, the double bond holds all 4 carbon atoms in the same plane as the atoms of the benzene ring, which as mentioned earlier is very important for receptor binding. The proposed phenethylamine/amphetamine 5HT2A agonists in fig 6 have the whole phenethyl skeleton held in the same plane due to the conjugation of the pi electrons with the delocalised electrons of the benzene ring, as well as presenting the pi electrons in the correct position. They should be more potent than their corresponding amphetamine derivative, as they will bond more strongly to the receptor. This also removes any complications that might occur with any adrenergic receptor interaction (the beta-hydroxy 2C-D derivative, BOHD caused a large drop in blood pressure – in fact the drug methoxamine has the 2,5-dimethoxy configuration, and a beta-hydroxy group, and it is a potent pressor agent), as the beta-hydroxy oxygen atom is in the same position as the benzylic hydroxy group oxygen atom. The corresponding DOM derivative would be 3-amino-2-(2,5-dimethoxy-4-methylphenyl)butene, and the 2C-D derivative would be 3-amino-2-(2,5-dimethoxy-4-methylphenyl)propene.
Also shown in fig 6 is a modification that retains the double bond, substituted into a tryptamine, but retaining the conjugated system of the aromatic nucleus (indole instead of benzene). This would produce a series of 4-vinyl-N, N-dialkyltryptamines, which again should be more potent than their corresponding 4-hydroxy-N,N-dialkyltryptamine, and as the group is far less polar, so should allow better penetration of the blood brain barrier
Fig 6a shows all these modifications taken to their logical conclusion in the double bond version of the fully aromatic dragonfly molecule. If in this molecule, there is an increase in potency of the same magnitude as would be expected when applied to DOB etc, then it could possibly be a phenethylamine derivative that is on a par with LSD in terms of binding, dose etc
Numbering system of phenethylamines, tryptamines and ergolines is outlined in the jpeg attachment
If anybody can be bothered to plough though all of that, please point if I’ve not seen some obvious flaw in my reasoning
Papers referenced
James J. Chambers, Deborah M. Kurrasch-Orbaugh, Matthew A. Parker, and David E. Nichols. Enantiospecific Synthesis and Pharmacological Evaluation of a Series of
Super-Potent, Conformationally Restricted 5-HT2A/2C Receptor Agonists. J. Med. Chem. 2001, 44, 1003-1010
Nicholas M. Barnes; Trevor Sharp. A review of central 5-HT receptors and their function. Neuropharmacology 38 (1999) 1083–1152
David E. Nichols, Ph.D. The Medicinal Chemistry of Phenethylamine Psychedelics. The Heffter Review of Psychedelic Research, Volume 1, 1998
F. A. B. Aldous, B. C. Barrass, K. Brewster, D. A. Buxton, D. M. Green, R. M. Pinder, P. Rich, M. Skeels, and K. J. Tutt. Structure-Activity Relationships in Psychotomimetic Phenylalkylamines. J.Med. Chem., 2974, Vol. 27, No. 20 1100 - 1111
Nichols D.E, LSD and Its Lysergamide Cousins. The Heffter Review of Psychedelic Research, Volume 2, 2001, 80-87
Shulgin Ann & Alexander T. PIHKAL (Phenethyamines I Have Known And Loved)
-----------------------------------------------------------------------------------------
The final bit was just theoretical pondering on what should happen, but it’ll really need somebody to synthesize it and carry out trials with it to definitely confirm or deny the activity
Attachments
Last edited:

