I've been out of the loop for awhile, but some of the Vargas results (e.g. lipophilicity correlates with psychoplastogenicity, membrane permeability is required for psychedelic-induced neuroplasticity) don't seem incompatible with the Moliner paper. Perhaps this allosteric TRKB site can only be accessed via an intracellular path?
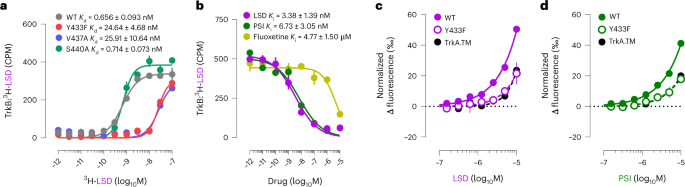
What is weird is that Vargas showed that ketanserin blocks both DMT and psilocin-mediated spinogenesis, while Moliner found that that M100907 has no effect on LSD-mediated spinogenesis. Moliner suggests that off-target effects of ketanserin (it has
high affinity for alpha-1 receptors and moderate affinity for alpha-2 receptors, among others) and not its 5-HT2A antagonism might account for its plasticity-inhibiting effects.
I wonder if a downstream consequence of this TRKB PAM activity is Gas redistribution away from lipid rafts and increased cAMP levels, which is something observed in vitro with both
ketamine,
HNK, and
fluoxetine, all of which share a common upstream mechanism (TRKB PAM).
Postmortem brain samples of depressed patients show increased Gas localization to lipid rafts, while
PET studies of depressed patients reveal global decreases in cAMP signaling which rebounds to control levels following antidepressant treatment (the Gas redistribution increases its functional coupling with adenylyl cyclase, which in turn increases cAMP levels). There's also potential for a positive feedback loop, as cAMP-PKA-CREB axis increases BDNF transcription (and
chronic fluoxetine increases BDNF mRNA).
I'd love to see some data on whether psychedelics can produce a similar redistribution of Gas (perhaps this is how
DOM and 25CN-NBOH increase cAMP levels in vitro), because I imaginee that ketanserin's off-target effects could interfere somewhere upstream of that. Btw, Lisuride was found to not increase cAMP, yet it does seem to be acting as a TRKB PAM. Very confusing..
Personally, in the case of ketamine, I think this TRKB PAM action probably isn't very relevant. It has even lower TRKB affinity than its metabolite HNK, yet
HNK plasma levels are negatively associated with antidepressant response to ketamine. Ketamine has a completely separate mechanism (inhibition of spontaneous NMDAr transmission) which has been studied in great detail, and that seems to be the main driver of its antidepressant activity.
 www.nature.com
www.nature.com


 but I just learned a thing or two so thank you for that.
but I just learned a thing or two so thank you for that.