MyDoorsAreOpen
Bluelight Crew
- Joined
- Aug 20, 2003
- Messages
- 8,549
Good point. The kind of vasodilators I had in mind are gingko and vincopetine which are believed to produce their nootropic effects, in part by enhancing blood flow to the brain. I need to research this more.
Ah, I see what you're saying. I don't know a whole lot about their mechanisms of action, but I have heard this said about a lot of substances used as nootropics. If you read more about what precise hematological changes ginkgo and others cause, definitely let me know!
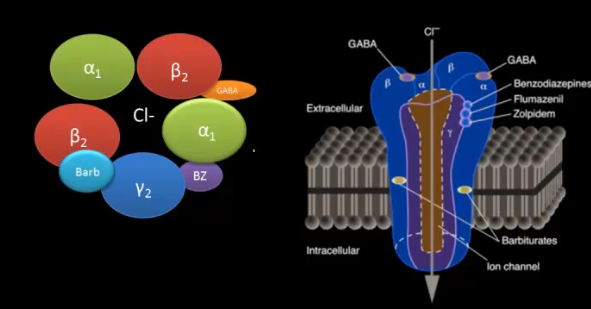
From what I read, its caused by the GABA_a antagonism induced by fluoroquinolones. All the reports of this I've heard, were with ciprofloxacin. I don't know whether its caused by glutamate excitotoxicity, or damage to the GABA neurons or what. BTW I meant to say taking a GABA_a direct agonist should help prevent this problem. Benzos on the other hand might make it worse, but I'm not sure. BZ agonists are positive allosteric modulators of the GABA_a receptor, so I wonder if that means they'd allow GABA_a antagonists to bind even more effectively to the GABA_a receptor.
GABA antagonism is seldom a good thing, because one of the major roles of GABAergic pathways is a safety valve for neurons firing too much and burning out. I've never given anyone flumazenil, and hope to never have to.
What you describe is not exactly what "allosteric binding" means. What it means is that the molecule has an affinity for a different spot on the receptor than the endogenous ligand our body produces to press that receptor, but sets off the same downstream cascade nonetheless. Believe it or not, benzos, barbituates, and ethanol all stick to different spots on the GABA receptor, and none of these are the same spot that GABA sticks to. So your answer your question, if a GABA antagonist's mode of action was to block GABA from binding to its receptor, then the presence of an allosteric ligand would have no bearing on the antagonist's binding affinity. However (and I suspect this is what you're driving at), so long as the shape and the way the antagonist adhered didn't block the benzodiazepine binding site also, then benzos would effectively protect against the damaging effects of unwanted GABA antagonism, so long serum levels were maintained until the antagonist was pretty much entirely out of the system.
I can't be bothered to look up the Ki of ciprofloxacin to the GABA-a receptor, but I can't imagine the binding affinity is very strong in most people, or this drug would never have made it to clinical trials, never mind to market. If there are some people who possess mutations in the genes for GABA-a receptors that allow them to bind cipro much more readily, it would be a worthwhile research project to identify these people. Their medical charts could be marked as people who should ideally never get this drug, or if they ever absolutely need it, must be given benzos concurrently. These little micro-differences in cell surface proteins are the future of medicine regimens tailored specifically to you.