dopamimetic
Bluelighter
- Joined
- Mar 21, 2013
- Messages
- 2,070
NADPH oxidase mediates depressive behavior induced by chronic stress in mice.
Stress is a potent risk factor for depression, yet the underlying mechanism is not clearly understood. In the present study, we explored the mechanism of development and maintenance of depression in a stress-induced animal model. Mice restrained for 2 h daily for 14 d showed distinct depressive behavior, and the altered behavior persisted for >3 months in the absence of intervention. Acute restraint induced a surge of oxidative stress in the brain, and stress-induced oxidative stress progressively increased with repetition of stress. In vitro, the stress hormone glucocorticoid generated superoxide via upregulation of NADPH oxidase. Consistently, repeated restraints increased the expression of the key subunits of NADPH oxidase, p47phox and p67phox, in the brain. Moreover, stressed brains markedly upregulated the expression of p47phox to weak restress evoked in the poststress period, and this molecular response was reminiscent of amplified ROS surge to restress. Pharmacological inhibition of NADPH oxidase by apocynin during the stress or poststress period completely blocked depressive behavior. Consistently, heterozygous p47phox knock-out mice (p47phox(+/-)) or molecular inhibition of p47phox with Lenti shRNA-p47phox in the hippocampus suppressed depressive behavior. These results suggest that repeated stress promotes depressive behavior through the upregulation of NADPH oxidase and the resultant metabolic oxidative stress, and that the inhibition of NADPH oxidase provides beneficial antidepression effects.
Involvement of NOX1/NADPH Oxidase in Morphine-Induced Analgesia and Tolerance
The involvement of reactive oxygen species (ROS) in morphine-induced analgesia and tolerance has been suggested, yet how and where ROS take part in these processes remains largely unknown. Here, we report a novel role for the superoxide-generating enzyme NOX1/NADPH oxidase in the regulation of analgesia and acute analgesic tolerance. In mice lacking Nox1 (Nox1−/Y), the magnitude of the analgesia induced by morphine was significantly augmented. More importantly, analgesic tolerance induced by repeated administration of morphine was significantly suppressed compared with that in the littermates, wild-type Nox1+/Y. In a membrane fraction obtained from the dorsal spinal cord, no difference was observed in morphine-induced [35S]GTPγS-binding between the genotypes, whereas morphine-stimulated GTPase activity was significantly attenuated in Nox1−/Y. At 2 h after morphine administration, a significant decline in [35S]GTPγS-binding was observed in Nox1+/Y but not in Nox1−/Y. No difference in the maximal binding and affinity of [3H]DAMGO was observed between the genotypes, but the translocation of protein kinase C isoforms to the membrane fraction following morphine administration was almost completely abolished in Nox1−/Y. Finally, the phosphorylation of RGS9-2 and formation of a complex by Gαi2/RGS9-2 with 14-3-3 found in morphine-treated Nox1+/Y were significantly suppressed in Nox1−/Y. Together, these results suggest that NOX1/NADPH oxidase attenuates the pharmacological effects of opioids by regulating GTPase activity and the phosphorylation of RGS9-2 by protein kinase C. NOX1/NADPH oxidase may thus be a novel target for the development of adjuvant therapy to retain the beneficial effects of morphine.
--
NOX has several homologs, including NOX2 implicated in neurodegenerative and psychiatric disorders (Sorce and Krause, 2009). NOX2 was also reported to take part in the development of neuropathic pain induced by nerve injury (Kim et al., 2010). NOX1 is not well understood, and although its role in some organs is being elucidated (Matsuno et al., 2005, Cui et al., 2011), its function in the nervous system is still unclear. Previously, we demonstrated the involvement of NOX1 in hyperalgesia using mice lacking the Nox1 gene (...)
(Free full text!)
Could explain somewhat why the NMDA antagonism approach to prevent tolerance development (to stimulants, opioids) did not work as expected for many, or at least eventually one begins to become tolerant to the NMDA antagonist itself. While it is disputed (afaik) whether the NMDA receptors do up regulate to a relevant degree at all with low-to-moderate antagonism, there undoubtedly is the phenomenon of dissociative tolerance.
And this dissociative tolerance could be mediated by upregulated NOX, leading through a cascade of mechanisms amongst other nasty effects also to a generally higher amount of glutamatergic neurotransmission (and thus higher doses of NMDA antagonists required for the same effects, as well as a general shift in effects because the other glutamate receptors are affected too- this includes partially positive reactions like the Ketamine-mediated rapid antidepressant effect, as well as negative ones ranging from excitatory rebound up to psychosis - and/or 'dissociative addiction').
Maybe the oxidative stress itself can lead to production of quinolinic acid - and things will escalate.
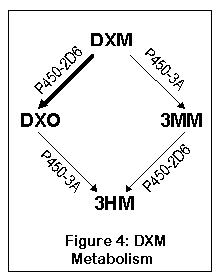
Now we have a readily available NOX inhibitor: dextromethorphan. Indeed it seems not only to exhibit qualitatively unique anxiolytic, antidepressant and especially stress-alleviating effects, but also lowers tolerance to NMDA antagonists and potentiates the anti-tolerance effects of e.g. memantine as proven by many anecdotal reports and myself - with DXM dosages used that are too low to act on NMDA etc. to a significant degree.
Apocynin is a vanillin-related compound that would be free of DXM's other effects and side effects. Unfortunately it's not available as a medicine yet, but it certainly needs to be further explored.
(As a side theory - since dopamine gets partially metabolized to homovanillic acid - which is structurally similar to apocynin - could it be that elevated dopamine levels will cause a downstream decrease of NADPH oxidase, and low DA levels potentially the opposite - leading to a novel explanation for the beneficial effects psychostimulants exhibit for e.g. ADHD even after one becomes used / tolerant to the acute effects? Besides the interesting fact that the 'non-stimulant' atomoxetine indeed is a NMDA antagonist in clinically relevant dosages- which probably mediates a good part of it's benefits.)
And we have that weird bupropion with it's insignificant NDRI properties, that looks like a somewhat locked-down cathinone that from time to time counters even parallel administered psychostimulants and indeed it modulates glutamate. In rats, it inhibits glutamate release. But rats also self-administer bupropion much more than humans, speculated due to differing metabolism.. now it depends if this inhibition is caused by the bupropion itself or dependent on the metabolism. (By accident I've discovered that DXM is able to 'unlock' bupropion, which now finally makes sense - with just 50mg of time-released DXM being enough to get powerful cathinone-like stimulation out of 150mg bupropion SR - but only when used chronically, not acutely. The CYP inhibition probably contributes to that, but I'd bet it's about NOX & glutamate, maybe as well as the SSRI effect from DXM, eventually leading to a triple reuptake inhibitor)
Just wild guessing, but it's somewhat intriguing to me
")
--
The NADPH oxidase NOX2 controls glutamate release: a novel mechanism involved in psychosis-like ketamine responses.
Ketamine-induced loss of phenotype of fast-spiking interneurons is mediated by NADPH-oxidase.
--
Edit: Last but not least, what negative / potentially dangerous effects could inhibiting the NOX have (acutely and chronically)? I highly guess that, as usual, there's nothing without a catch and these superoxides are necessary for something like immune response ...
Last edited:

 so it's still in there. If I clench my jaw, the left tinnitus indeed changes the pitch somewhat, yes.. but if anything then the DXM or NMDA antagonism in general is more muscle relaxing than otherwise. No more tension headaches! And memantine has been a muscle relaxant in the end before they re-patented it for Alzheimer's and decided to make another billion shitload of $$$ with it.
so it's still in there. If I clench my jaw, the left tinnitus indeed changes the pitch somewhat, yes.. but if anything then the DXM or NMDA antagonism in general is more muscle relaxing than otherwise. No more tension headaches! And memantine has been a muscle relaxant in the end before they re-patented it for Alzheimer's and decided to make another billion shitload of $$$ with it.