why they use ketamine to anesthesize the animals before drug administration (see paper)??. Isnt ketamine and/or its metabolites potentially interfere with the effect of ecstasy?? or the metabolism of DXM? but it looks convincing the protective effet of DXM on ecstasy induce neurotoxity
Effects of dextromethorphan on MDMA-induced serotonergic aberration in the brains of non-human primates using [123I]-ADAM/SPECT; Ma et al. Sci Rep. 2016; 6: 38695. Published online 2016 Dec 12. doi: 10.1038/srep38695
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5150522/
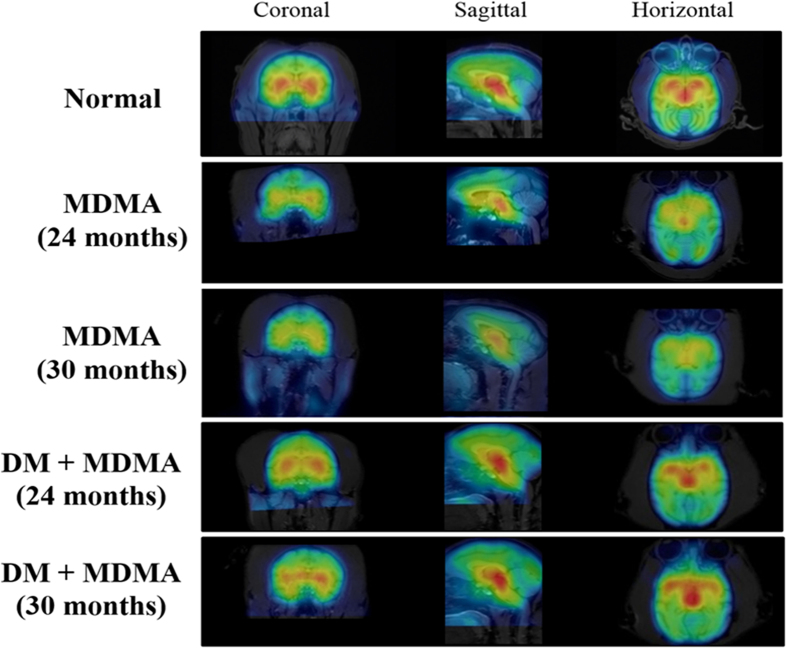
ABSTRACT: 3,4-Methylenedioxymethamphetamine (MDMA), a common recreational drug, is known to cause serotonergic neurotoxicity in the brain. Dextromethorphan (DM) is a widely used antitussive reported to exert anti-inflammatory effect in vivo. In this study, we examined the long-term effect of MDMA on the primate serotonergic system and the protective property of DM against MDMA-induced serotonergic abnormality using single photon emission computed tomography (SPECT). Nine monkeys (Macaca cyclopis) were divided into three groups, namely control, MDMA and co-treatment (MDMA/DM). [123I]-ADAM was used as the radioligand for serotonin transporters (SERT) in SPECT scans. SERT levels of the brain were evaluated and presented as the uptake ratios (URs) of [123I]-ADAM in several regions of interest of the brain including midbrain, thalamus and striatum. We found that the URs of [123I]-ADAM were significantly lower in the brains of MDMA than control group, indicating lower brain SERT levels in the MDMA-treated monkeys. This MDMA-induced decrease in brain SERT levels could persist for over four years. However, the loss of brain SERT levels was not observed in co-treatment group. These results suggest that DM may exert a protective effect against MDMA-induced serotonergic toxicity in the brains of the non-human primate.
") just because they can't tell their buddies or write their experience on erowid or BL doesnt mean they didn't have that same experience as "humans".. but who knows?
just because they can't tell their buddies or write their experience on erowid or BL doesnt mean they didn't have that same experience as "humans".. but who knows?